
Abstract
In this review, we examine the evidence that intestinal helminths can control harmful inflammatory responses and promote homeostasis by triggering systemic immune responses. Induction of separable components of immunity by helminths, which includes type 2 and immune regulatory responses, can both contribute toward the reduction in harmful type 1 immune responses that drive certain inflammatory diseases. Despite inducing type 2 responses, intestinal helminths may also downregulate harmful type 2 immune responses including allergic responses. We consider the possibility that intestinal helminth infection may indirectly affect inflammation by influencing the composition of the intestinal microbiome. Taken together, the studies reviewed herein suggest that intestinal helminth-induced responses have potent systemic effects on the immune system, raising the possibility that whole parasites or specific molecules produced by these metazoans may be an important resource for the development of future immunotherapies to control inflammatory diseases.
Similar content being viewed by others
INTRODUCTION
Approximately two billion people are currently infected with intestinal helminths, with the heaviest worm burdens occurring in non-industrialized countries.1 This group of parasites can be soil transmitted, such as with hookworms (Necator and Ancylostoma), whipworms (Trichuris), roundworms (Ascaris), or tapeworms (Cestoda). Other intestinal helminths, for example, Trichinella, can also be transmitted through oral ingestion of larval-infected tissues, such as by eating undercooked meats. Intestinal helminths can impair fitness in both wild2, 3 and domestic4, 5, 6 mammalian hosts. In some cases, infection with intestinal helminths can impair physical and cognitive development in children.7 It is thus likely that these ubiquitous helminths have exerted considerable selection pressure during the several hundred million years they have coevolved with their mammalian hosts.8, 9 The evolution of the type 2 immune response is a likely consequence of the selection pressure from helminths, although other potential elements driving the evolution of the type 2 immune response have been proposed.9, 10, 11
The type 2 response is characterized by increased production by immune cells of T helper type 2 (Th2) cytokines, including: interleukin (IL)-4, IL-5, IL-9, and IL-13. Although initial studies identified CD4+ Th2 cells as an important source of these cytokines, it is now clear that a number of other immune cell lineages, including eosinophils, basophils, and innate lymphoid cells (ILCs) can be activated to produce one or more of these cytokines during helminth infection.12, 13, 14, 15, 16, 17, 18, 19 As such, these cytokines will be referred to as type 2 cytokines. The immune regulatory cytokine IL-10 was initially described as a Th2 cytokine; however, it is now clear that IL-10 is also made by Th1 cells and various types of Treg cells. Even during helminth infection expression of IL-10 can be regulated independently of the type 2 response.20, 21, 22, 23, 24 Another important regulatory cytokine induced by helminth infection is transforming growth factor beta (TGF-β). TGF-β is also regulated independently of type 2 cytokines, and can be produced by Treg cells.25, 26, 27
It is useful to think of the immune response evoked by helminths as composed of two separable and at times independent compartments, a type 2 cytokine response and a regulatory response, both of which contribute to the overall protective type 2 response to helminth parasites. Generally, this overall protective response includes mechanisms that mediate resistance, resulting in accelerated worm expulsion, and also mechanisms that enhance tolerance of invasive helminths. In this context, we refer to tolerance as the ability to reduce damage to the host without affecting resistance to the pathogen.28, 29 In the host response to intestinal helminths, tolerance includes both direct effects promoting wound healing and also inhibitory effects on harmful inflammation.9, 30, 31, 32 This harmful inflammation is frequently mediated by the type 1 immune response and can be defined as including the elevation of both Th1 and Th17 cytokines, as previously described.9, 29
Macrophages may also mediate immune regulatory effects through expression of arginase-1, PDL1, and PDL2,33 IL-10,34 and Socs-1.35 Recent studies suggest that during acute stages of helminth infection, IL-10 is predominantly produced by T cells and not by macrophages.30, 36 Hence, macrophages during these acute responses have an alternatively activated macrophage (M2) phenotype, which is more consistent with a wound healing rather than a regulatory phenotype, as recently described.37 However, M2 macrophages have also been shown to induce the differentiation of Tregs through their production of retinoic acid.38 Overall, the acute type 2 response is important in enabling tolerance by the host of the considerable tissue damage caused by intestinal helminths during their life cycle, including migration through primary organs such as the lung, as in the case of hookworms. Taken together, the mammalian host both resists and tolerates helminth infections through the evolution of complementary components of type 2 and regulatory responses.9 However, it should also be noted that prolonged type 2 immunity, particularly in chronic responses, can also lead to immune pathology, including fibrosis.39, 40
INTESTINAL HELMINTH-INDUCED RESPONSES
The intestinal immune response may have been the first component of the adaptive immune response to evolve in vertebrates.41, 42 This response must distinguish nutrients and their associated antigens from potential pathogens. It must also distinguish commensal and mutualistic symbionts, which may be vital components of the microbiome, from harmful microbial pathogens. Additionally, there is the need to distinguish between pathogenic microbes and eukaryotic multicellular parasites, to trigger the appropriate type 1 or type 2 immune response. An area of active investigation is to understand how the mucosal immune response manages the complex task of recognition and activation of appropriate immune effector cell populations.
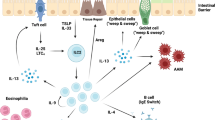
Considerable progress has recently been made in determining the molecular triggers that initiate both type 1 and type 2 mucosal immune responses in the enteric milieu. In the case of helminths, increases in type 2 cytokines, in particular IL-5 and IL-13, can be detectable within hours after infection. An important source of these cytokines is innate lymphoid 2 cells (ILC-2s). ILC-2s release type 2 cytokines following activation by cytokine alarmins, including IL-25, IL-33, and thymic stromal lymphopoietin. These alarmins are rapidly released by endothelial and epithelial cells,13, 14, 15, 43 and likely other cell types,44, 45, 46, 47, 48 at early stages of the response. Biologically active IL-33 is released by necrotic cells during tissue damage,49 thereby providing a link between the type 2 response and tissue injury. Mast cells activated during helminth infection can also promote cytokine alarmin production in some cases playing an essential role in promoting the type 2 immune response.50
Intestinal epithelial cells form the initial barrier between the lumen and intestinal tissues and may be a primary source for cytokine alarmin production. The lamina propria supports a robust population of immune cells, which are rapidly stimulated following intestinal nematode infection (Figure 1). Alarmin activated ILC-2s produce primarily IL-5 and IL-13, while activation of eosinophils and basophils may serve as a primary initial source of IL-4.51, 52, 53, 54 Thus, these granulocytes may preferentially support Th2 cell differentiation.55 Recent studies suggest that ILC-2s may actually activate eosinophils, through their production of IL-5, thereby indirectly triggering IL-4 release.56 Activated dendritic cells (DCs) and other innate immune cell populations migrate from the lamina propria, and the more organized peyers patches, through lymphatic vessels to the mesenteric lymph node. Here, a potent adaptive immune response, which includes Th2 cells, and B cells secreting IgE and IgG1, soon develops during helminth infection of mice. The mesenteric lymph node is likely an important source of the elevated serum IgE and IgG1 detected within 2 weeks after Heligmosomoides polygyrus bakeri inoculation, when germinal center formation is readily detected.57

Intestinal helminths initially trigger a type 2 immune response through multiple mechanisms. Helminths damage tissue during feeding and migration in the intestine, which in turn can trigger the release of danger-associated molecular patterns (DAMPs) and cytokine alarmins (interleukin (IL)-25, thymic stromal lymphopoietin (TSLP), and IL-33). These alarmins then activate innate lymphoid cells and likely other cell populations that support the development of a type 2 innate immune response, which in turn provides necessary signals for the development of type 2 adaptive immunity. Excretory/secretory (ES) products released by helminths also promote the type 2 immune response through stimulation of cytokine alarmins and through effects on dendritic cells (DCs) that inhibit their capacity to support T helper type 1 (Th1) cell differentiation while promoting Th2 cell and T regulatory cell development in the draining lymph nodes. At the site of infection, the type 2 immune microenvironment likely sustains activation of Th2 effector cell and T regulatory cells. Together tissue damage and ES products, in the absence of strong Toll-like receptor (TLR) signaling, are likely primary stimuli that together trigger and help maintain a robust multicomponent type 2 response. Effects of helminths on the microbiome may also influence mucosal and systemic inflammatory responses. TGF-β, transforming growth factor beta.
The spleen may also be an important source of serum Igs and potentially Th2 cells during the response to H. polygyrus bakeri, which is a strictly enteric helminth (Figure 2). Splenomegaly is detected by 2 weeks after inoculation,58 which is associated with increased type 2 cytokine gene expression in the spleen,59, 60, 61, 62 indicating that a systemic response rapidly follows the initial mucosal response after intestinal helminth infection. Use of bicistronic 4get IL-4 reporter mice has demonstrated accumulation of Th2 cells in the liver, lung airways, and the peritoneal cavity after H. polygyrus bakeri inoculation. Thus, Th2 cells themselves spread systemically during this strictly enteric nematode parasite infection.60 As might be expected, however, the enteric response does initially develop before the systemic response and is associated with higher levels of Th2 cytokine expression.60, 63 Studies of wild wood mice (Apodemus sylvaticus) have further shown a negative correlation between splenocyte responsiveness to Toll-like receptor signaling and chronic infection with H. polygyrus bakeri,64 indicating that intestinal helminth infection may systemically downregulate microbial recognition and host responsiveness in wild rodent populations.

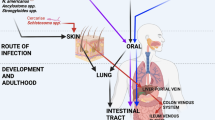
Intestinal helminths may stimulate systemic responses through lymphatic/circulatory system. Intestinal helminths trigger a potent type 2 innate immune response in the gut that stimulates activated dendritic cells (DCs) to migrate through the lymphatic ducts to the draining mesenteric lymph node (mLN) where they can promote T helper 2 (Th2) cell differentiation. At the same time, helminth excretory/secretory (ES) products may also enter the circulation and contribute to the development of adaptive type 2 immune responses. Dissemination of ES products and activated cells through the circulation to other organs may contribute to the development of systemic responses and increases in Th2 cells in liver, spleen, and peritoneum. Thus, an immune response induced by helminths in the enteric region may have broad systemic anti-inflammatory effects. Intestinal helminths that migrate from the skin through the vasculature to the lung and then the intestine also induce localized responses at these additional sites that can contribute to systemic immune responses. IL, interleukin; TGF-β, transforming growth factor beta; IGF-1, insulin-like growth factor 1.
While activating type 2 responses, specific helminth excretory/secretory (ES) products can have the capacity to inhibit antigen-presenting cells, predominantly DCs, from producing IL-12, costimulatory molecules, and other factors required for Th1 cell differentiation.65, 66, 67, 68, 69, 70, 71 Instead, helminth-activated DCs may stimulate the development of T regulatory populations that further downregulate type 1 responses.69, 71, 72, 73, 74 This cascade of effects results in the development of two at least partially independent and complementary immune responses to helminths, the first resulting in cytokine alarmin release and stimulation of type 2 cytokines and the second resulting in the development of DC populations that stimulate Treg cell activation and downregulate type 1 cytokines. Together, these different activation cascades can promote a potent and polarized type 2 immune response accompanied by strong immunoregulatory populations.
INTESTINAL HELMINTH MIGRATION TRIGGERS POTENT NON-ENTERIC IMMUNE RESPONSES
The migration of larval stages can also extend the effects of intestinal helminths on immune function far beyond the site of infection. There are several experimental rodent models of helminth infection where the immune response is initiated entirely in the intestinal milieu including H. polygyrus, Trichuris muris, and Trichinella spiralis.75 A well-characterized rodent model of helminth infection with systemic larval migration is the nematode parasite Nippostrongylus brasiliensis (Figure 2), which like the human hookworm, infects through the skin and undergoes a lung migratory phase. This migration from the site of infection in the skin through the lungs and finally to the intestine can have pronounced effects on systemic immune responses, even after the parasites are no longer present in the affected tissues.76, 77, 78, 79, 80, 81 In the lungs a lymphoid infiltrate occurs within 48 h after hookworm larval invasion, which can adversely affect lung function when given at high doses in experimental models.30, 78 A rapid influx of neutrophils and associated tissue damage is controlled by an acute type 2 immune response. Pronounced elevations in IL-4 and IL-13 control harmful inflammation at least in part through activation of M2 macrophages. Macrophage-derived factors, including insulin-like growth factor 1 beta, directly promote tissue repair while other factors, including arginase-1, mediate control of inflammation. Hence, the severity of acute lung injury is reduced through an IL-4R-dependent mechanism.30 Even after the parasites have exited the lungs and entered the small intestine, a chronic type 2 response in the lung is sustained. The chronic type 2 response can eventually lead to long-term lung damage that can include emphysematous pathology.82, 83
Interestingly, a secondary challenge with N. brasiliensis triggers accelerated worm expulsion, with most parasites killed by several days after challenge, usually before larvae reach the intestine. Both the skin and the lungs are likely sites of immune attack by macrophages and other immune cell populations, resulting in worm killing.79, 84 The potent pulmonary immune response to migrating N. brasiliensis larvae may also trigger increased susceptibility to infection with Mycobacterium tuberculosis. This could at least in part be due to the alternative activation of macrophages in the lung by the type 2 immune response, resulting in impaired macrophage bactericidal effector functions.85 As recently reviewed,86 increasing evidence suggests that in humans, as well as in experimental models, the immune response to intestinal helminths may influence protective mucosal and systemic immune responses to microbial pathogens, including human immune deficiency virus, Plasmodium species that cause malaria, and M. tuberculosis.
INTESTINAL HELMINTHS CONTROL HARMFUL INFLAMMATION BEYOND THE INTESTINAL MUCOSA
The potent systemic effect of an intestinal helminth infection is further demonstrated by the effective prevention of spontaneous type 1 diabetes (T1D) in non-obese diabetic (NOD) mice following H. polygyrus inoculation59, 61, 87 (Figure 3). NOD mice develop an aggressive autoimmune response toward the islet beta cells of the pancreas, resulting in peri-insulitis at about 5–6 weeks of age followed by invasive insulitis, destruction of the islet beta cells, and development of diabetes as early as 12–14 weeks of age.88, 89 H. polygyrus inoculation effectively prevents invasive insulitis and associated diabetes when administered at 5 weeks of age and even significantly reduces the incidence of T1D when administered as late as 7 weeks of age.59, 87 Furthermore, if H. polygyrus is administered at 5 weeks of age and then worms are expulsed by drug treatment at 7 weeks, development of T1D is still blocked as late as 40 weeks of age.61 Thus, only a 2-week interval of enteric helminth infection is needed to block the development of T1D. This potent effect on T1D, where only a short time window is required for the presence of Th2 stimulating agents, raises the possibility of future effective protocols utilizing short-term intestinal helminth-based therapies for the control of T1D in humans. Intriguingly, even when IL-4R signaling is essentially blocked in NOD-IL-4−/− mice, effective blockade of T1D still occurs following H. polygyrus inoculation.61 Similarly, T1D is also prevented in NOD-IL-4−/− mice after systemic infection with the filarial parasitic nematode Litosomoides sigmondontis. In the L. sigmondontis model, TGF-β was essential for control of T1D in the absence of IL-4 and for the associated blockade of IL-13 and other type 2 cytokines.90 Thus, potent immune regulatory mechanisms, occurring independently of type 2 cytokines, are induced by nematode parasites after systemic or intestinal helminth infection. Further studies with H. polygyrus showed that loss of either IL-4 or IL-10 signaling alone was not sufficient to abrogate the protective effects of helminth infection against the formation of T1D while combined blockade of IL-4 and IL-10 lead to loss of H. polygyrus-mediated control of invasive insulitis.61

Intestinal helminths trigger immune regulatory cells that can inhibit type1 diabetes and associated inflammation. Intestinal helminth infection stimulates the development of a potent type 2 immune response in the pancreatic lymph associated with decreased islet beta cell-specific interferon gamma (IFN-γ) producing Th1 cells and an increase in interleukin (IL)-4, transforming growth factor beta (TGF-β), and auto antigen-specific T cells producing IL-10. We propose that this may occur because the helminth-induced immune microenvironment in this draining lymph node alters dendritic cell (DC) activation such that these auto-antigen presenting cells now favor the development of regulatory Tr1 cells instead of inflammatory Th1 cells. We propose that this effect dampens the stimulus provided by IFN-γ that would otherwise promote the development of an inflammatory response leading to invasive insulitis and destruction of islet beta cells. It should be noted that in this context the spleen may also be an important source regulatory cell populations and cytokines.
These findings provide insight into why the helminth-induced type 2 immune response is so robust, as it apparently includes immune regulatory compartments that can develop independently and are at least partly redundant. An important source of the IL-10 in the H. polygyrus-inoculated NOD mice was FoxP3−, CD4+ autoantigen-specific T cells,61 which is consistent with a Tr1 (type 1 regulatory cell) phenotype. In one model, Tr1 cells originate from conventional interferon gamma (IFN-γ) producing Th1 cells, with an intermediate Th cell producing both IL-10 and IFN-γ.20, 22 In the context of T1D, such helminth-induced differentiation of islet beta cell autoreactive IFN-γ producing Th1 to IL-10 producing Tr1 cells would not only provide an important source of immunomodulatory IL-10 but would also effectively deplete the major source of IFN-γ, which has an essential role in the development of invasive insulitis.91, 92, 93, 94, 95
It is perhaps surprising that a strictly intestinal nematode parasite can have such potent effects on a systemic autoimmune disease. This may simply be a result of potent helminth-derived stimuli spreading through the lymphatics to the circulatory system eventually affecting the spleen and other secondary lymphoid tissues including the pancreatic lymph node (see Figure 3). There is, however, also recent evidence suggesting that islet-reactive T cells in NOD mice initially develop in the mesenteric lymph nodes and express gut homing receptors including MAdCAM-1 and α4β7, raising the possibility that these autoreactive T cells may be cross reactive with gut-derived foreign Ags.96, 97 This apparent association of the development of T1D with the enteric response may thus be a factor in the potent control of T1D by intestinal helminth-induced responses, particularly at early stages in the development of the autoimmune response. Interestingly, systemic administration of ES-62, a potent helminth-derived anti-inflammatory product, which is effective in treating experimental allergy98 and arthritis models,99 has little effect on arresting the development of T1D in NOD mice (Harnett and Cooke, personal communication).
Intestinal helminth infections may also suppress symptoms of allergic responses. Superficially, this observation may seem paradoxical, as both helminths and allergies activate type 2 immune responses. One possible explanation may be that, in contrast to the allergic response, the immune response to helminths includes additional immune regulatory components, at least partly triggered by ES products from the worms.100 ES products may attenuate DC activation by suppressing upregulation of costimulatory molecules and cytokines.69 As already discussed, ES-activated DCs can support the development of Treg populations, resulting in the upregulation of immunoregulatory cytokines including IL-10. As well as dampening Th2 cell function, recent studies suggest that IL-10 produced during chronic helminth infection may also directly modulate allergic effector cells, specifically basophils, by reducing their responsiveness to IgE-mediated stimulation.101 Thus, ES-activated DCs may control allergic inflammation during helminth infection and potentially attenuate responses to unrelated allergens.
Multiple clinical studies have suggested that allergic diseases are more pronounced in areas of low helminth endemicity.102, 103, 104, 105, 106, 107 In one large study, children infected with geohelminths, including Ascaris lumbricoides (ascariasis), Trichuris trichiura (trichuriasis), and Ancylostoma duodenale, showed increased protection against allergen skin test reactivity.108 Although the mechanisms remain unclear, this is likely mediated through multiple regulatory components, as elevations in type 2 response markers (e.g., serum IgE), but not IL-10, were associated with increased control of atopy in A. lumbricoides-infected children.109
Studies with mouse models have also demonstrated modulatory effects of intestinal helminth infections on allergic responses. In particular, H. polygyrus infection can control experimental airway induced allergic responses through IL-10-independent mechanisms, likely involving Treg cells.110 Recent studies have further shown that H. polygyrus derived ES products alone are sufficient to effectively inhibit experimental allergic airway inflammation.100 One possible mechanism of ES regulatory effects may be through TGF-β signaling, as H. polygyrus derived ES does include a TGF-β mimic.73 However, in this airway model exogenous TGF-β administration had little effect and, unlike TGF-β, H. polygyrus derived ES was heat stable.100 Intriguingly, ES products from H. polygyrus bakeri may also directly inhibit IL-33 release that would otherwise promote innate type 2 immune responses contributing to airway allergy.111
These data support the hypothesis that helminths and their ES products likely operate through multiple mechanisms. These could either be redundant mechanisms, or interact through either additive or synergistic mechanisms to enhance immunomodulatory effects. This model may help explain why helminth-induced modulatory effects are both robust and potent. It should also be noted that, although live helminths likely induce stronger responses than ES products alone,9 individual ES components or combinations thereof may still be sufficient and effective in the development of future therapies to treat harmful type 1 or type 2 inflammatory responses.
HELMINTH INFECTION AND THE METABOLIC SYNDROME
Recent studies have implicated eosinophils and M2 macrophages in maintaining metabolic homeostasis.112, 113 In contrast, classically activated (M1) macrophages, characteristic of the type 1 immune response, produce inflammatory cytokines, tumor necrosis factor-α and IL-6, which can decrease insulin sensitivity. Along with their production of resistin, M1 macrophages can thus potentially mediate insulin resistance and glucose intolerance, thereby contributing to metabolic syndrome and the development of obesity and type 2 diabetes.114, 115, 116 M2 macrophages, activated by IL-4 and IL-13, do not make tumor necrosis factor-α or IL-6 and respond to signaling through the receptor peroxisome proliferator-activated receptor-α, which instead dampens expression of these inflammatory cytokines. Eosinophils, residing in adipose tissue, produce IL-4, which drives M2 macrophage differentiation.112 Furthermore, recent studies suggest that ILC-2s may be an important early source of IL-5, which then promotes eosinophil IL-4 production.56 As helminth infection upregulates expression of IL-4 and other type 2 cytokines, it is perhaps not surprising that N. brasiliensis infection of mice fed a high calorie diet markedly reduces both adipose tissue mass and the likelihood of developing glucose intolerance.108 Intriguingly, infection with even the strictly enteric pathogen, H. polygyrus, can also modulate energy homeostasis.117 These are more examples of the potent effects of intestinal helminth infection on shaping systemic responses. Like many other inflammatory diseases, the prevalence of metabolic syndrome is increasing in industrialized countries which are less exposed to gastrointestinal helminth infections, thus raising the possibility that their absence may be a contributing factor to systemic inflammatory disease pathogenesis.113
HELMINTHS AND THE MICROBIOME
Although helminths can directly modulate the immune system (as described above), it is also possible that they may indirectly influence the immune response through their effects on the intestinal microbiome.118 Changes in intestinal commensal bacteria have been implicated in the altered immunity that can result in inflammatory diseases both in the localized intestinal milieu119, 120 and systemically.121, 122, 123 Chronic intestinal helminth infections have recently been shown to affect the composition of the microbiome. Chronic infection with H. polygyrus was shown to favor specific types of commensal bacteria, in particular members of the Lactobacillaceae family.124 Studies with T. muris indicate that intestinal microbiota provide environmental cues that can facilitate helminth egg hatching,125 suggesting complex interactions between intestinal helminths and resident microbes. Recent studies also suggest that fecal microbial communities in developed and undeveloped countries are markedly different; in particular, the genus Prevotella is more common in developed countries.126 This may result in part from dietary differences, but the prevalence of helminth infections may also be a factor.118
IL-22, which is increased in humans and mice by intestinal helminth infection,127 may function as a cytokine mediator between the microbiome and the intestinal tissue.118 By promoting mucous production, IL-22 facilitates tissue repair and mucosal barrier formation.128, 129, 130 IL-22 can also enhance production of anti-microbial peptides, including β-defensin family proteins.131, 132, 133, 134 IL-22-deficient mice showed increased systemic dissemination of intestinal commensal bacteria to the spleen and liver,135 raising the possibility that helminth-induced IL-22 production may contribute to control of inflammation during inflammatory bowel disease by containing bacteria within the intestinal lumen. However, studies with S. mansoni-infected mice have shown little effect of IL-22 deficiency on intestinal or liver granulomas.136 It will be interesting in future studies to examine a possible role for intestinal helminth infection in maintaining the mucosal barrier137 and also in reversing dysbiosis,118 two potentially interacting mechanisms that may contribute to the control of inflammatory diseases.
CLINICAL STUDIES
The success in several experimental models using intestinal helminth infection or ES administration to modulate harmful type 1 and type 2 systemic and mucosal inflammatory responses provides a basis for future clinical study of their effects on autoimmune and idiopathic inflammatory diseases. There have now been a few clinical trials using helminths to treat patients. Trichuris suis ova (TSO) has been used in phase I clinical trials to treat Crohn’s disease.138, 139 The results were sufficiently encouraging that much larger phase II clinical trials were undertaken. However, these trials appear to have been unsuccessful, indicating that a further understanding of heterogeneity in systemic responses to helminths is required. The initial studies showed evidence for increases in blood eosinophilia for some patients, indicative of a systemic response to TSO. As Trichuris suis is not a native human pathogen, this strictly enteric parasite resides only transiently in the human host. How TSO triggers systemic eosinophilia and whether this is associated with a polarized type 2 response that is analogous to the response induced by a native human helminth will need to be examined.
Promising results were found in another small trial in which TSO was used to treat multiple sclerosis. Using magnetic resonance imaging, lesions were found to be decreased after 3 months of TSO treatment, but by 2 months after cessation of treatment they were again increased.140 Furthermore, recent trials with TSO for allergic rhinitis have not shown a therapeutic benefit,141 although it has been suggested that there may have been confounding factors including limited dosing, timing of treatment, and differences between active and placebo-treated groups in use of medications and baseline symptoms.142, 143, 144 Initial safety trials have also used hookworm infections as a potential allergy treatment, suggesting an alternative approach that would include direct stimulation of lung immunity by migrating parasites.144 There is certainly the need for more extensive and robust clinical trials, which incorporate mechanistic measurements of responses.
In each of the human diseases where treatment with TSO or other parasite preparations may be considered, useful biomarkers will need to be identified that allow tracking of the potency, polarization, and systemic dissemination of the helminth-elicited response. This may include general indicators of type 2 responses, such as elevations in serum IgE, eosinophilia, and increases in Th2 cytokines, including IL-4 and IL-13. In cases where Ag-specific responses associated with the harmful inflammatory response are known, for example, T1D, it will be useful to assess the Ag-specific response and in the case of Th cells, interrogate the Th1/Th2/Th17 cytokine pattern. Unfortunately, none of these types of analyses were undertaken in the recently failed phase II trials of TSO for Crohn’s disease; hence, it is unclear as to whether the trials failed because TSO did not elicit a strong response in these patients, or whether there was considerable heterogeneity in responses to TSO.
CONCLUSIONS
The type 2 immune response, which is potently triggered by intestinal helminths, is important in mammalian physiology in terms of host defense, wound healing, and tissue homeostasis. Even strictly enteric helminths can induce potent systemic responses that can dampen type 1 immune responses that would otherwise cause harmful inflammatory responses. Further understanding of how this type 2 immune response works and what characteristics of helminths actually trigger it is required to exploit its potential as a platform for developing new treatments for a broad group of inflammatory diseases. Specific questions that need to be addressed include: (i) Are there other as yet undiscovered immunoregulatory cell types or molecules associated with helminth infection? (ii) Can individual ES products or combinations thereof substitute effectively for helminths in mediating control of inflammatory responses? (iii) To what extent is the intestinal microbiome influenced by helminth infection and do these effects in turn influence the systemic immune response? (iv) Are some systemic inflammatory responses refractory or more sensitive to effects of helminth infection? Although helminths appear to have a generalized dampening effect on harmful inflammatory responses, subtle differences including localization and accessibility to regulatory immune cells may be important. (v) Under what conditions might harmful aspects of the type 2 immune response, such as fibrosis, limit the use of this response for targeted therapies? Overall, the potency and relative specificity of the multicomponent type 2 immune response to modulate harmful type 1 responses provides an exciting opportunity for the development of new treatments for inflammatory responses contributing to a wide range of diseases.
References
de Silva, N.R. et al. Soil-transmitted helminth infections: updating the global picture. Trends Parasitol. 19, 547–551 (2003).
Gulland, F.M. The role of nematode parasites in Soay sheep (Ovis aries L.) mortality during a population crash. Parasitology 105 (Pt 3), 493–503 (1992).
Gulland, F.M. & Fox, M. Epidemiology of nematode infections of Soay sheep (Ovis aries L.) on St Kilda. Parasitology 105 (Pt 3), 481–492 (1992).
Li, R.W., Choudhary, R.K., Capuco, A.V. & Urban, J.F. Jr. Exploring the host transcriptome for mechanisms underlying protective immunity and resistance to nematode infections in ruminants. Vet. Parasitol. 190, 1–11 (2012).
Charlier, J., Hoglund, J., von Samson-Himmelstjerna, G., Dorny, P. & Vercruysse, J. Gastrointestinal nematode infections in adult dairy cattle: impact on production, diagnosis and control. Vet. Parasitol. 164, 70–79 (2009).
Valentine, B.A., Cebra, C.K. & Taylor, G.H. Fatal gastrointestinal parasitism in goats: 31 cases (2001–2006). J. Am. Vet. Med. Assoc. 231, 1098–1103 (2007).
King, C.H. Health metrics for helminthic infections. Adv. Parasitol. 73, 51–69 (2010).
Jackson, J.A., Friberg, I.M., Little, S. & Bradley, J.E. Review series on helminths, immune modulation and the hygiene hypothesis: immunity against helminths and immunological phenomena in modern human populations: coevolutionary legacies? Immunology 126, 18–27 (2009).
Gause, W.C., Wynn, T.A. & Allen, J.E. Type 2 immunity and wound healing: evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol. 13, 607–614 (2013).
Palm, N.W., Rosenstein, R.K. & Medzhitov, R. Allergic host defences. Nature 484, 465–472 (2012).
Artis, D., Maizels, R.M. & Finkelman, F.D. Forum: Immunology: allergy challenged. Nature 484, 458–459 (2012).
Liang, H.E. et al. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat. Immunol. 13, 58–66 (2012).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Saenz, S.A. et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366 (2010).
Neill, D.R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Wills-Karp, M. & Finkelman, F.D. Innate lymphoid cells wield a double-edged sword. Nat. Immunol. 12, 1025–1027 (2011).
Voehringer, D., Reese, T.A., Huang, X., Shinkai, K. & Locksley, R.M. Type 2 immunity is controlled by IL-4/IL-13 expression in hematopoietic non-eosinophil cells of the innate immune system. J. Exp Med 203, 1435–1446 (2006).
van Panhuys, N. et al. Basophils are the major producers of IL-4 during primary helminth infection. J. Immunol. 186, 2719–2728 (2011).
Rosenberg, H.F., Dyer, K.D. & Foster, P.S. Eosinophils: changing perspectives in health and disease. Nat. Rev. Immunol. 13, 9–22 (2013).
Anderson, C.F., Oukka, M., Kuchroo, V.J. & Sacks, D. CD4(+)CD25(−)Foxp3(−) Th1 cells are the source of IL-10-mediated immune suppression in chronic cutaneous leishmaniasis. J. Exp. Med. 204, 285–297 (2007).
Jankovic, D. et al. Conventional T-bet(+)Foxp3(−) Th1 cells are the major source of host-protective regulatory IL-10 during intracellular protozoan infection. J. Exp. Med. 204, 273–283 (2007).
Cope, A., Le Friec, G., Cardone, J. & Kemper, C. The Th1 life cycle: molecular control of IFN-gamma to IL-10 switching. Trends Immunol. 32, 278–286 (2011).
Hang, L. et al. Heligmosomoides polygyrus bakeri infection activates colonic Foxp3+ T cells enhancing their capacity to prevent colitis. J. Immunol. 191, 1927–1934 (2013).
Rubtsov, Y.P. et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008).
Reynolds, L.A. & Maizels, R.M. Cutting edge: in the absence of TGF-beta signaling in T cells, fewer CD103+ regulatory T cells develop, but exuberant IFN-gamma production renders mice more susceptible to helminth infection. J. Immunol. 189, 1113–1117 (2012).
Reynolds, L.A., Filbey, K.J. & Maizels, R.M. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin. Immunopathol. 34, 829–846 (2012).
Hubner, M.P. et al. Helminth protection against autoimmune diabetes in nonobese diabetic mice is independent of a type 2 immune shift and requires TGF-beta. J. Immunol. 188, 559–568 (2012).
Schneider, D.S. & Ayres, J.S. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8, 889–895 (2008).
Medzhitov, R., Schneider, D.S. & Soares, M.P. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Chen, F. et al. An essential role for T(H)2-type responses in limiting acute tissue damage during experimental helminth infection. Nat. Med. 18, 260–266 (2012).
Pesce, J.T. et al. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 5, e1000371 (2009).
Herbert, D.R. et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. J. Immunol. 184, 6438–6446 (2010).
Loke, P. & Allison, J.P. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc. Natl. Acad. Sci. USA 100, 5336–5341 (2003).
de Waal Malefyt, R., Abrams, J., Bennett, B., Figdor, C.G. & de Vries, J.E. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J. Exp. Med. 174, 1209–1220 (1991).
Whyte, C.S. et al. Suppressor of cytokine signaling (SOCS)1 is a key determinant of differential macrophage activation and function. J. Leukoc. Biol. 90, 845–854 (2011).
Thomas, G.D. et al. The biology of nematode- and IL4Ralpha-dependent murine macrophage polarization in vivo as defined by RNA-Seq and targeted lipidomics. Blood 120, e93–e104 (2012).
Mosser, D.M. & Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969 (2008).
Broadhurst, M.J. et al. Upregulation of retinal dehydrogenase 2 in alternatively activated macrophages during retinoid-dependent type-2 immunity to helminth infection in mice. PLoS Pathog. 8, e1002883 (2012).
Duffield, J.S., Lupher, M., Thannickal, V.J. & Wynn, T.A. Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 (2013).
Borthwick, L.A., Wynn, T.A. & Fisher, A.J. Cytokine mediated tissue fibrosis. Biochim. Biophys. Acta 1832, 1049–1060 (2013).
Lee, Y.K. & Mazmanian, S.K. Has the microbiota played a critical role in the evolution of the adaptive immune system? Science 330, 1768–1773 (2010).
Matsunaga, T. & Rahman, A. In search of the origin of the thymus: the thymus and GALT may be evolutionarily related. Scand. J. Immunol. 53, 1–6 (2001).
Saenz, S.A. et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J. Exp. Med. 210, 1823–1837 (2013).
Siracusa, M.C. et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 477, 229–233 (2011).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490 (2005).
Chang, Y.J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 12, 631–638 (2011).
Ikeda, K. et al. Mast cells produce interleukin-25 upon Fc epsilon RI-mediated activation. Blood 101, 3594–3596 (2003).
Fock, V. et al. Macrophage-derived IL-33 is a critical factor for placental growth. J. Immunol. 191, 3734–3743 (2013).
Oboki, K., Nakae, S., Matsumoto, K. & Saito, H. IL-33 and Airway Inflammation. Allergy Asthma Immunol. Res. 3, 81–88 (2011).
Hepworth, M.R. et al. Mast cells orchestrate type 2 immunity to helminths through regulation of tissue-derived cytokines. Proc. Natl. Acad. Sci. USA 109, 6644–6649 (2012).
Khodoun, M.V., Orekhova, T., Potter, C., Morris, S. & Finkelman, F.D. Basophils initiate IL-4 production during a memory T-dependent response. J. Exp. Med. 200, 857–870 (2004).
Mitre, E., Taylor, R.T., Kubofcik, J. & Nutman, T.B. Parasite antigen-driven basophils are a major source of IL-4 in human filarial infections. J. Immunol. 172, 2439–2445 (2004).
Gessner, A., Mohrs, K. & Mohrs, M. Mast cells, basophils, and eosinophils acquire constitutive IL-4 and IL-13 transcripts during lineage differentiation that are sufficient for rapid cytokine production. J. Immunol. 174, 1063–1072 (2005).
Voehringer, D., Shinkai, K. & Locksley, R.M. Type 2 immunity reflects orchestrated recruitment of cells committed to IL-4 production. Immunity 20, 267–277 (2004).
Hung, L.Y. et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proc. Natl. Acad. Sci. USA 110, 282–287 (2013).
Nussbaum, J.C. et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013).
Ekkens, M.J. et al. Memory Th2 effector cells can develop in the absence of B7-1/B7-2, CD28 interactions, and effector Th cells after priming with an intestinal nematode parasite. J. Immunol. 168, 6344–6351 (2002).
Brailsford, T.J. & Mapes, C.J. Comparisons of Heligmosomoides polygyrus primary infection in protein-deficient and well-nourished mice. Parasitology 95 (Pt 2), 311–321 (1987).
Liu, Q. et al. Helminth infection can reduce insulitis and type 1 diabetes through CD25- and IL-10-independent mechanisms. Infect. Immun. 77, 5347–5358 (2009).
Mohrs, K., Harris, D.P., Lund, F.E. & Mohrs, M. Systemic dissemination and persistence of Th2 and type 2 cells in response to infection with a strictly enteric nematode parasite. J. Immunol. 175, 5306–5313 (2005).
Mishra, P.K., Patel, N., Wu, W., Bleich, D. & Gause, W.C. Prevention of type 1 diabetes through infection with an intestinal nematode parasite requires IL-10 in the absence of a Th2-type response. Mucosal Immunol. 6, 297–308 (2013).
Perona-Wright, G., Mohrs, K. & Mohrs, M. Sustained signaling by canonical helper T cell cytokines throughout the reactive lymph node. Nat. Immunol. 11, 520–526 (2010).
Svetic, A. et al. Cytokine gene expression after in vivo primary immunization with goat antibody to mouse IgD antibody. J. Immunol. 147, 2391–2397 (1991).
Friberg, I.M. et al. Macroparasites at peripheral sites of infection are major and dynamic modifiers of systemic antimicrobial pattern recognition responses. Mol Ecol 22, 2810–2826 (2013).
Massacand, J.C. et al. Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc. Natl. Acad. Sci. USA 106, 13968–13973 (2009).
Al-Riyami, L. & Harnett, W. Immunomodulatory properties of ES-62, a phosphorylcholine-containing glycoprotein secreted by Acanthocheilonema viteae. Endocr. Metab. Immune Disord. Drug Targets 12, 45–52 (2012).
Chaussabel, D. et al. Unique gene expression profiles of human macrophages and dendritic cells to phylogenetically distinct parasites. Blood 102, 672–681 (2003).
Kane, C.M. et al. Helminth antigens modulate TLR-initiated dendritic cell activation. J. Immunol. 173, 7454–7461 (2004).
Segura, M., Su, Z., Piccirillo, C. & Stevenson, M.M. Impairment of dendritic cell function by excretory-secretory products: a potential mechanism for nematode-induced immunosuppression. Eur. J. Immunol. 37, 1887–1904 (2007).
Everts, B. et al. Schistosome-derived omega-1 drives Th2 polarization by suppressing protein synthesis following internalization by the mannose receptor. J. Exp. Med. 209, 1753–1767 (2012).
McSorley, H.J., Hewitson, J.P. & Maizels, R.M. Immunomodulation by helminth parasites: defining mechanisms and mediators. Int. J. Parasitol. 43, 301–310 (2013).
Aranzamendi, C. et al. Trichinella spiralis-secreted products modulate DC functionality and expand regulatory T cells in vitro. Parasite Immunol. 34, 210–223 (2012).
Grainger, J.R. et al. Helminth secretions induce de novo T cell Foxp3 expression and regulatory function through the TGF-beta pathway. J Exp Med 207, 2331–2341 (2010).
Hartmann, W., Haben, I., Fleischer, B. & Breloer, M. Pathogenic nematodes suppress humoral responses to third-party antigens in vivo by IL-10-mediated interference with Th cell function. J. Immunol. 187, 4088–4099 (2011).
Patel, N., Kreider, T., Urban, J.F. Jr. & Gause, W.C. Characterisation of effector mechanisms at the host:parasite interface during the immune response to tissue-dwelling intestinal nematode parasites. Int. J. Parasitol. 39, 13–21 (2009).
Daly, C.M., Mayrhofer, G. & Dent, L.A. Trapping and immobilization of Nippostrongylus brasiliensis larvae at the site of inoculation in primary infections of interleukin-5 transgenic mice. Infect. Immun. 67, 5315–5323 (1999).
Knott, M.L. et al. Impaired resistance in early secondary Nippostrongylus brasiliensis infections in mice with defective eosinophilopoeisis. Int. J. Parasitol. 37, 1367–1378 (2007).
Harvie, M. et al. The lung is an important site for priming CD4 T-cell-mediated protective immunity against gastrointestinal helminth parasites. Infect. Immun. 78, 3753–3762 (2010).
Harvie, M., Camberis, M. & Le Gros, G. Development of CD4 T cell dependent immunity against N. brasiliensis infection. Front. Immunol 4, 74 (2013).
Herrick, C.A., Xu, L., McKenzie, A.N., Tigelaar, R.E. & Bottomly, K. IL-13 is necessary, not simply sufficient, for epicutaneously induced Th2 responses to soluble protein antigen. J. Immunol. 170, 2488–2495 (2003).
Liu, Z. et al. Nippostrongylus brasiliensis can induce B7-independent antigen-specific development of IL-4-producing T cells from naive CD4 T cells in vivo. J. Immunol. 169, 6959–6968 (2002).
Marsland, B.J., Kurrer, M., Reissmann, R., Harris, N.L. & Kopf, M. Nippostrongylus brasiliensis infection leads to the development of emphysema associated with the induction of alternatively activated macrophages. Eur. J. Immunol. 38, 479–488 (2008).
Heitmann, L. et al. TGF-beta-responsive myeloid cells suppress type 2 immunity and emphysematous pathology after hookworm infection. Am. J. Pathol. 181, 897–906 (2012).
Obata-Ninomiya, K. et al. The skin is an important bulwark of acquired immunity against intestinal helminths. J. Exp. Med. 210, 2583–2595 (2013).
Potian, J.A. et al. Preexisting helminth infection induces inhibition of innate pulmonary anti-tuberculosis defense by engaging the IL-4 receptor pathway. J. Exp Med 208, 1863–1874 (2011).
Salgame, P., Yap, G.S. & Gause, W.C. Effect of helminth-induced immunity on infections with microbial pathogens. Nat. Immunol. 14, 1118–1126 (2013).
Saunders, K.A., Raine, T., Cooke, A. & Lawrence, C.E. Inhibition of autoimmune type 1 diabetes by gastrointestinal helminth infection. Infect. Immun. 75, 397–407 (2007).
Anderson, M.S. & Bluestone, J.A. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 23, 447–485 (2005).
Tisch, R. & McDevitt, H. Insulin-dependent diabetes mellitus. Cell 85, 291–297 (1996).
Hubner, M.P. et al. Helminth protection against autoimmune diabetes in nonobese diabetic mice is independent of a type 2 immune shift and requires TGF-beta. J. Immunol. 188, 559–568 (2012).
Suk, K. et al. IFN-gamma/TNF-alpha synergism as the final effector in autoimmune diabetes: a key role for STAT1/IFN regulatory factor-1 pathway in pancreatic beta cell death. J. Immunol. 166, 4481–4489 (2001).
Trembleau, S., Penna, G., Gregori, S., Giarratana, N. & Adorini, L. IL-12 administration accelerates autoimmune diabetes in both wild-type and IFN-gamma-deficient nonobese diabetic mice, revealing pathogenic and protective effects of IL-12-induced IFN-gamma. J. Immunol. 170, 5491–5501 (2003).
Esensten, J.H., Lee, M.R., Glimcher, L.H. & Bluestone, J.A. T-bet-deficient NOD mice are protected from diabetes due to defects in both T cell and innate immune system function. J. Immunol. 183, 75–82 (2009).
Callewaert, H.I. et al. Deletion of STAT-1 pancreatic islets protects against streptozotocin-induced diabetes and early graft failure but not against late rejection. Diabetes 56, 2169–2173 (2007).
Lehuen, A., Diana, J., Zaccone, P. & Cooke, A. Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 10, 501–513 (2010).
Hanninen, A., Salmi, M., Simell, O. & Jalkanen, S. Mucosa-associated (beta 7-integrinhigh) lymphocytes accumulate early in the pancreas of NOD mice and show aberrant recirculation behavior. Diabetes 45, 1173–1180 (1996).
Jaakkola, I., Jalkanen, S. & Hanninen, A. Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur. J. Immunol. 33, 3255–3264 (2003).
Rzepecka, J. et al. The helminth product, ES-62, protects against airway inflammation by resetting the Th cell phenotype. Int. J. Parasitol. 43, 211–223 (2013).
Pineda, M.A. et al. The parasitic helminth product ES-62 suppresses pathogenesis in collagen-induced arthritis by targeting the interleukin-17-producing cellular network at multiple sites. Arthritis Rheum. 64, 3168–3178 (2012).
McSorley, H.J. et al. Suppression of type 2 immunity and allergic airway inflammation by secreted products of the helminth Heligmosomoides polygyrus. Eur. J. Immunol. 42, 2667–2682 (2012).
Larson, D. et al. Chronic helminth infection reduces basophil responsiveness in an IL-10-dependent manner. J. Immunol. 188, 4188–4199 (2012).
Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 347, 911–920 (2002).
Asher, M.I. et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet 368, 733–743 (2006).
Cooper, P.J. Interactions between helminth parasites and allergy. Curr. Opin. Allergy Clin. Immunol. 9, 29–37 (2009).
Hagel, I. et al. Modulation of the allergic reactivity of slum children by helminthic infection. Parasite Immunol. 15, 311–315 (1993).
Nyan, O.A. et al. Atopy, intestinal helminth infection and total serum IgE in rural and urban adult Gambian communities. Clin. Exp. Allergy 31, 1672–1678 (2001).
Scrivener, S. et al. Independent effects of intestinal parasite infection and domestic allergen exposure on risk of wheeze in Ethiopia: a nested case-control study. Lancet 358, 1493–1499 (2001).
Cooper, P.J. et al. Reduced risk of atopy among school-age children infected with geohelminth parasites in a rural area of the tropics. J. Allergy Clin. Immunol. 111, 995–1000 (2003).
Cooper, P.J. et al. Ascaris lumbricoides-induced interleukin-10 is not associated with atopy in schoolchildren in a rural area of the tropics. J. Infect. Dis 197, 1333–1340 (2008).
Wilson, M.S. et al. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J. Exp. Med. 202, 1199–1212 (2005).
McSorley, H.J., Blair, N.F., Smith, K.A., McKenzie, A.N. & Maizels, R.M. Blockade of IL-33 release and suppression of type 2 innate lymphoid cell responses by helminth secreted products in airway allergy. Mucosal Immunol. advance online publication, 5 February 2014; doi:10.1038/mi.2013.123 (e-pub ahead of print).
Wu, D. et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011).
Maizels, R.M. & Allen, J.E. Immunology. Eosinophils forestall obesity. Science 332, 186–187 (2011).
Weisberg, S.P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 (2003).
Muse, E.D. et al. Role of resistin in diet-induced hepatic insulin resistance. J. Clin. Invest. 114, 232–239 (2004).
Olefsky, J.M. & Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev.Physiol 72, 219–246 (2010).
Wong, T. et al. Divergent metabolic adaptations to intestinal parasitic nematode infection in mice susceptible or resistant to obesity. Gastroenterology 133, 1979–1988 (2007).
Leung, J.M. & Loke, P. A role for IL-22 in the relationship between intestinal helminths, gut microbiota and mucosal immunity. Int. J. Parasitol. 43, 253–257 (2013).
Devkota, S. & Chang, E.B. Nutrition, microbiomes, and intestinal inflammation. Curr. Opin Gastroenterol. 29, 603–607 (2013).
Strober, W. Impact of the gut microbiome on mucosal inflammation. Trends Immunol. 34, 423–430 (2013).
Brusca, S.B., Abramson, S.B. & Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 26, 101–107 (2013).
Vinje, S., Stroes, E., Nieuwdorp, M. & Hazen, S.L. The gut microbiome as novel cardio-metabolic target: the time has come!. Eur. Heart J. advance online publication, 11 November 2013; doi:10.1093/eurheartj/eht467(e-pub ahead of print).
Naseer, M.I. et al. Role of gut microbiota in obesity, type 2 diabetes and Alzheimer’s disease. CNS Neurol. Disord. Drug Targets advance online publication, 18 September 2013 (e-pub ahead of print).
Walk, S.T., Blum, A.M., Ewing, S.A., Weinstock, J.V. & Young, V.B. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm. Bowel Dis. 16, 1841–1849 (2010).
Hayes, K.S. et al. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 328, 1391–1394 (2010).
De Filippo, C. et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 107, 14691–14696 (2010).
Broadhurst, M.J. et al. IL-22+ CD4+ T cells are associated with therapeutic trichuris trichiura infection in an ulcerative colitis patient. Sci. Transl. Med. 2, 60ra88 (2010).
Sonnenberg, G.F., Fouser, L.A. & Artis, D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 12, 383–390 (2011).
Sonnenberg, G.F. et al. Pathological versus protective functions of IL-22 in airway inflammation are regulated by IL-17A. J. Exp. Med. 207, 1293–1305 (2010).
Turner, J.E., Stockinger, B. & Helmby, H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog. 9, e1003698 (2013).
Zenewicz, L.A. & Flavell, R.A. Recent advances in IL-22 biology. Int. Immunol. 23, 159–163 (2011).
Wolk, K. et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur. J. Immunol. 36, 1309–1323 (2006).
Zindl, C.L. et al. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 110, 12768–12773 (2013).
Liang, S.C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 (2006).
Sonnenberg, G.F. et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336, 1321–1325 (2012).
Wilson, M.S. et al. Redundant and pathogenic roles for IL-22 in mycobacterial, protozoan, and helminth infections. J. Immunol. 184, 4378–4390 (2010).
Wolff, M.J., Broadhurst, M.J. & Loke, P. Helminthic therapy: improving mucosal barrier function. Trends Parasitol. 28, 187–194 (2012).
Summers, R.W., Elliott, D.E., Urban, J.F. Jr., Thompson, R. & Weinstock, J.V. Trichuris suis therapy in Crohn's disease. Gut 54, 87–90 (2005).
Summers, R.W. et al. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am. J. Gastroenterol. 98, 2034–2041 (2003).
Fleming, J.O. et al. Probiotic helminth administration in relapsing-remitting multiple sclerosis: a phase 1 study. Mult. Scler. 17, 743–754 (2011).
Bager, P. et al. Trichuris suis ova therapy for allergic rhinitis: a randomized, double-blind, placebo-controlled clinical trial. J. Allergy Clin. Immunol. 125, 123–130 e121–123 (2010).
Summers, R.W., Elliott, D.E. & Weinstock, J.V. Trichuris suis might be effective in treating allergic rhinitis. J. Allergy Clin. Immunol. 125, 766–767 (2010).
Hepworth, M.R., Hamelmann, E., Lucius, R. & Hartmann, S. Looking into the future of Trichuris suis therapy. J. Allergy Clin. Immunol. 125, 767–768 author reply 768-769 (2010).
Pritchard, D.I., Blount, D.G., Schmid-Grendelmeier, P. & Till, S.J. Parasitic worm therapy for allergy: is this incongruous or avant-garde medicine? Clin. Exp. Allergy 42, 505–512 (2012).
Acknowledgements
We acknowledge Sevier medical art for their templates in www.servier.com and figures were produced using those templates. This work was in part supported by NIH grants 1R01AI107588 and 5R01AI031678 awarded to WCG and NIH grants R01 AI093811 and NIH/NCATS UL1 TR000038 awarded to P.L. In addition, P.L. is supported by The Broad Medical Research Program; Kevin and Marsha Keating Family Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
P.L. collaborates with Coronado Biosciences and has served Ad Hoc on their Scientific Advisory Panel.
Rights and permissions
About this article

Cite this article
Mishra, P., Palma, M., Bleich, D. et al. Systemic impact of intestinal helminth infections. Mucosal Immunol 7, 753–762 (2014). https://doi.org/10.1038/mi.2014.23
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2014.23
This article is cited by
-
Systemic profile of immune factors in an elderly Italian population affected by chronic strongyloidiasis
Parasites & Vectors (2020)
-
Virtual memory CD8 T cells expanded by helminth infection confer broad protection against bacterial infection
Mucosal Immunology (2019)
-
Metabolic adaptations of tissue-resident immune cells
Nature Immunology (2019)
-
Chronic helminth infection burden differentially affects haematopoietic cell development while ageing selectively impairs adaptive responses to infection
Scientific Reports (2018)
-
Immune modulation of Th1, Th2, and T-reg transcriptional factors differing from cytokine levels in Schistosoma japonicum infection
Parasitology Research (2018)
