
Abstract
Chronic inflammation has been associated with increased risk for developing gastrointestinal cancer. Interleukin-23 (IL-23) receptor signaling has been correlated with inflammatory bowel disease pathogenesis, as well as promotion of tumor growth. However, little is known about the relative potential for IL-23-directed causality in gut tumorigenesis. We report that IL-23 transgene expression was sufficient to induce rapid (3–4 weeks) de novo development of intestinal adenomas with 100% incidence. Initiation of tumorigenesis was independent of exogenous carcinogens, Helicobacter colonization, or pre-existing tumor-suppressor gene mutations. Tumorigenesis was mediated by Thy1+IL-23R+ innate lymphoid cells (ILC3), in part, through IL-17 responses as tumor development was inhibited in RAG−/− × IL-17−/− double knockout mice. Remarkably, IL-23 initiation of tumorigenesis by resident ILCs consistently occurred before recruitment of conspicuous inflammatory infiltrates. Our results reveal an explicit role for IL-23-mediated initiation of gut tumorigenesis and implicate a key role for IL-23R+ ILC3 in the absence of overt cellular infiltrate recruitment.
Similar content being viewed by others


Introduction
The association of inflammation and gastrointestinal cancer is now well established.1 Although bacterial factors can contribute to oncogenesis, most evidence suggests that host factors are paramount in determining tumor progression. Upregulation of pro-inflammatory cytokines associated with anti-bacterial immune responses, particularly in the chronic inflammation setting, have been shown to be contributing factors to tumorigenesis.2 In particular, numerous reports have demonstrated an association of interleukin (IL)-1β, IL-6, and tumor necrosis factor-α (TNFα) and IL-11 responses to increase cancer risk and tumor progression.1, 3, 4, 5, 6
More recently, the IL-23/IL-17 cytokine regulatory axis has been shown to have an essential role in immune-mediated inflammatory diseases.7, 8 Importantly, genome-wide association studies in inflammatory bowel disease (IBD) patient cohorts identified single-nucleotide polymorphisms within the IL-23 receptor (IL-23R) gene that support the hypothesis that IL-23 contributes significantly to the predisposition and pathogenesis of IBD,9, 10 in which there are well-documented increased risks for development of cancer in the colon and the upper gastrointestinal tract.11, 12, 13 In pre-clinical mouse models, IL-23 has been reported to promote carcinogen-induced tumorigenesis and tumor growth in transplantable tumor models, demonstrating that the IL-23 regulatory pathways contributed to tumor progression.14, 15 Recent reports have shown T helper type 17 (Th17) cells to be associated with increased adenoma development in APCMin models16, 17 and for the induction of gastroduodenal pre-malignant polyps in mice deficient for SMAD-4 signaling.18 The involvement of Th17 cells in promoting neoplastic transformation in the gut suggests that IL-23 may be a contributing upstream regulatory cytokine. IL-23 has been demonstrated to be a key inducer of pathogenic phenotypes mediated by Th17 cells.7 Importantly, recent evidence indicates that IL-23-associated gut pathogenesis is also induced by innate lymphoid cells (ILCs), mediated in part by IL-23/IL-17 cytokine regulatory pathways expressed by the ILCs.19, 20 Collectively, these reports demonstrate that the IL-23/IL-17 axis in inflammation is associated with tumor-promoting activity, which was secondary to tumor initiation first induced by chemical carcinogenesis, tumor suppressor-gene inactivation, or persistent infection with specific pathogenic bacterial strains. Our investigations focused on assessing the relative potential for IL-23-directed causality for initiating, as well as promoting, intestinal tumorigenesis and to elucidate the specific cellular and molecular responses associated with neoplastic transformation and progression. Pre-clinical studies of the specific role of IL-23 and the downstream responses on gut tumorigenesis have been hampered by the lack of viable IL-23 transgenic mouse models, which succumb to sequelae associated with severe systemic inflammation before reaching breeding age.21 We therefore utilized IL-23 minicircle (MC) hydrodynamic delivery (HDD) technology to induce stable long-term expression of IL-23 in adult mice to investigate the role of IL-23 cytokine regulation in gut tumorigenesis and to elucidate the mechanisms associated with downstream responses.
We report here that IL-23 transgene expression in wild-type (WT) mice was sufficient to drive rapid de novo development of gut adenomas with a virtually 100% incidence in the absence of any exogenously added carcinogen, colonization with Helicobacter hepaticus, or pre-existing tumor-suppressor gene mutations. Neoplastic events occur as early as 1 week following induction of IL-23 expression and development of macroscopic adenomas can be observed by 3 weeks. Surprisingly, early tumorigenesis commenced in the absence of overt inflammatory infiltrates or increases in lamina propria (LP) cell numbers. Adenomas arising from the villus epithelium required IL-23R signaling in lymphoid cells as tumorigenesis was completely inhibited in RAG−/− × IL-2Rγc−/− double knockout mice. Further characterization revealed a key role for IL-17-producing ILCs expressing Thy-1, Sca-1, and the IL-23R as essential initiating cells. These results support the premise that IL-23 is a key upstream regulatory cytokine, sufficient and essential for the induction of de novo gut tumorigenesis.
Results
Systemic IL-23 exposure induces duodenal adenomas
Previous attempts to produce IL-23p19 transgenic mice resulted in severe inflammation in multiple organs and mice incapable of breeding.21 We utilized MC DNA HDD technology to investigate the effects of chronic IL-23-mediated inflammation and tumorigenesis in the gut.22, 23 C57BL/6 mice, infused by HDD with IL-23 MC, developed adenomas in the proximal region of the duodenum immediately below the pyloric sphincter separating the stomach from the small intestine (Figure 1a–d). Multiple tumors were present at 150 days post IL-23 MC HDD (Figure 1c). Adenomas, pre-malignant tumors arising from the villus epithelium, developed in all the observed mice with sustained serum exposure of IL-23 >600 pg ml−1 (Figure 1e). The site of tumor development was remarkably consistent and only rarely extended into the pyloris. Adenoma development required IL-23R signaling as tumors were completely absent in IL-23R knockouts systemically expressing IL-23 (Figure 1f).

Systemic expression of interleukin (IL)-23 induces duodenal adenomas. Wild-type mice were injected with 3 μg of control minicircle (albumin) DNA or IL-23 DNA and killed 150 days post injection. (a) Gross and (b) hematoxylin and eosin (H&E)-sectioned duodenal tissue from control mice. (c) Gross and (d) H&E-sectioned duodenal tissue from IL-23 treated mice. Arrows indicate adenomas. Representative images (at least 3 mice per group) are shown. The dotted line in a and c marks the pyloric sphincter. (e) Required serum IL-23 expression for adenoma development. Various amounts of IL-23 minicircle DNA (0.003–3 μg) were injected to achieve a range of serum IL-23 expression. The dotted line denotes 0.6 ng ml−1. (n=33) (f) Adenoma formation in IL-23 receptor–deficient mice. In all, 3 μg of IL-23 minicircle DNA was injected into IL-23R−/− mice, and tumor development was assessed after 150 days (n=5). Bars indicate 100 μm (second column, b and d) and 50 μm (third column, b and d). HDD, hydrodynamic delivery.
Surprisingly, macroscopic tumors were observed as early as 3 weeks post IL-23 MC HDD induction (Figure 2a). At this time point, there were only one or two adenomas found (Figure 2b). Histopathological assessment revealed changes consistent with early neoplastic development that were evident in the proximal duodenum within 1 week of IL-23 expression; regions of villus malformation, villus loss, and loss of epithelial cell polarity were already apparent (Figure 2c compared with Figure 1b). At 2 and 3 weeks, these features along with structural disorganization became more prominent (Figure 2c). Remarkably, the LP appeared normal, and there was no evidence of overt increases in inflammatory infiltration present at these time points of early adenoma development.

Interleukin (IL)-23 systemic expression results in abnormal epithelial cell growth, neoplastic transformation and macroscopic tumor formation within 3 weeks of expression. Wild-type mice were injected with 2 μg IL-23 minicircle DNA and killed 1–4 weeks post injection. (a) Tumor development was quantified at the indicated time points post DNA hydrodynamic delivery (HDD). Representative data from two independent experiments, seven mice per group. (b) Representative image of the macroscopic tumors (arrows) evident at 3 weeks of expression from a. (c, d) Representative images of (c) hematoxylin and eosin and (d) Ki67 staining of abnormal duodenal tissue from a, scale bars indicate 200 (c) or 100 μm (d).
Staining for Ki67, a cell proliferative marker, revealed a progression of mitotic cells advancing upward from the crypts to the surface epithelium, providing further evidence for a loss of normal differentiation associated with neoplastic transformation. In control-treated proximal duodenal tissue, proliferating cells were all localized at the base of the villi in the intestinal crypts (Figure 2d). In contrast, in IL-23-expressing mice, proliferating cells extended beyond the crypt regions with replicating cells advancing further over time and from an increasing number of crypts (Figure 2d). At 3 weeks, proliferating cells were located throughout most of the adenomatous region (Figure 2d). These data indicated that systemic IL-23-regulated responses were sufficient for induction of dysregulated epithelial cell proliferation in the proximal duodenum that lead to adenoma formation.
The observation that IL-23 initiation of tumorigenesis occurred before explicit increases in inflammatory infiltrates was surprising. A previous report of cytokine initiation of gastric tumors in 1-year-old IL-1β transgenic mice24 was associated with extensive gastritis with inflammatory infitration. We utilized immunohistochemical (IHC) staining for CD3, F4/80, and myeloperoxidase (MPO) as a means to further investigate potential inflammatory infiltrates in the LP. As illustrated in Figure 3a, T-cell numbers and distribution, as evaluated by CD3 IHC, were similar in tumor-adjacent normal tissue and proximal duodenum from mice expressing control trangene DNA but were much reduced in the adenoma tissue itself. Macrophage lineage cells, as evaluated by F4/80 staining, were also similar in number and distribution between control and tumor-adjacent normal tissue but were rare to absent in the IL-23-induced adenomas. In contrast, MPO-positive cells, consistent with the presence of neutrophils, were present in low numbers in the superficial LP of adenomatous tissue but were rare to absent in control or tumor-adjacent normal tissue. Analysis of LP cells per mg of tissue in flow cytometry experiments revealed no differences in proximal duodenum of control and IL-23-expressing mice (Figure 3b), supporting the observation for a lack of overt inflammatory infiltrate associated with early IL-23 initiation of tumorigenesis compared with other models of inflammation-induced tumorigenesis.

Interleukin (IL-23) induces duodenal adenomas in the absence of overt inflammatory infiltrates. Wild-type mice were injected with control or IL-23 minicircle and killed 2 weeks post injection. Representative photomicrographs are shown from at least five mice per group. (a) Control proximal duodenal tissue, normal tumor-adjacent, and tumor tissue were stained for CD3, F4/80, and myeloperoxidase (MPO) expression by immunohistochemistry for histological examination. Arrows point to MPO+ cell clusters in tumor tissue. (b) Duodenal lamina propria (LP) cells from control or IL-23-expressing mice were isolated and normalized to starting amounts of tissue. Averages from three independent experiments are shown. Scale bars represent 200 and 50 μm (for an enlarged MPO image). NS, not significant.
Altered gene expression consistent with cell proliferation and tumor growth
Gene expression analysis was performed on tissues from the tumor initiation site at one week and and on tumors vs. normal tumor-adjacent (NTA) biopsy samples 4 and 21 weeks following IL-23 MC delivery. Results were compared with proximal duodenal tissues from albumin control MC HDD–injected mice. Remarkably, dysregulation of cell cycle gene expression was significantly increased for cyclin-dependent kinases (CDK2 and CDK4) and cyclin D (CCND1 and CCND3) at the tumor-initiation site in the proximal duodenum as early as 1 week following induction with IL-23. Furthermore, increased expression levels of CDKs and CCND was consistently observed in tumors but not in NTA tissue in early tumors (4 weeks) and throughout tumor progression (21 weeks) (Figure 4a, respectively). Likewise, expression of oncogenes and associated genes that regulate growth factor induced signaling, such as AKT (AKT1), nuclear factor-κB (NFκB; NFKB and RELA), and activator protein-1 (AP-1; FOS and JUNB), were upregulated as early as 1 week at the tumor initiation site and in early adenomas at 4 weeks. In more advanced 21-week tumors, upregulation was largely limited to the adenomas as compared with NTA tissue and control normal tissue (Figure 4b). Differential expression of AKT, NFκB, and AP-1 were not as evident for early tumors (4 weeks) and NTA tissue samples. Although a prospective mechanism for oncogene upregulation was not found, tumor-specific and IL-23-dependent upregulation of pro-inflammatory and wound-healing regulatory genes implicated as positive modulators of intestinal tumorigenesis, such as Cox-2, Myc, SOX9, IGFBP5, CXCL1, CD44, S100A8, MMP7, and REG3β, were observed (Figure 4a). Upregulation of TTF3, which is associated with intestinal tumor progression, was also evident. Although most of these genes were only significantly elevated in the tumor tissue, MMP7 and REG3β were also increased in the NTA tissue. These data taken together indicate a rapid and sustained induction of altered gene expression profile in IL-23-driven adenomas that is consistent with cellular proliferation and tumor growth progression.

Altered expression of genes associated with proliferation and tumor growth. Wild-type mice were injected with control or interleukin (IL)-23 minicircle and killed after (a) 1 or 4 weeks or (b) approximately 150 (21 weeks) days post injection. (a) RNA was isolated from proximal duodenal tissue and cDNA was prepared and analyzed by quantitative PCR; gene expression was normalized to ubiquitin. Averages and s.e.m. are shown (control n=3, 1 week n=5, 4 weeks n=5); NTA, normal tumor-adjacent tissue. Statistically significant differences compared with control (*) are indicated. *P<0.05; **P<0.01; ***P<0.001. (b) RNA was prepared from normal proximal duodenal tissue of control minicircle-injected mice (control), NTA, and tumor tissue (tumor) in IL-23 minicircle–injected mice, following 21 weeks of Tg expression. Gene expression analysis was performed as in a. Averages and s.e.m. are shown (control n=5, NTA tissue n=24, tumor n=27). Statistically significant differences compared with control (*) or with NTA tissue (#) are indicated. *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ###P<0.001. CCND1, cyclin D1; CDK2, cyclin-dependent kinase 2; COX2, cyclooxygenase-2; CXCL1, C-C motif chemokine ligand 1; IGFBP5, insulin-like growth factor–binding protein 5; MMP7, matrix metalloproteinase 7; NF, nuclear factor.
IL-23 initiation of epithelial tumorigenesis follows ILC activation as well as stimulation of IL-17 and interferon-γ (IFNγ) signaling pathways
Most reports show that the primary responsive cell types for IL-23 are comprised of hematopoietic-derived immune cells expressing IL-23R.25, 26, 27 We utilized IL-23R green fluorescent protein (GFP) reporter mice that have the intracellular domain of IL-23R replaced with GFP27 to identify IL-23R-expressing cells by GFP expression. Distinct GFP+ cells, isolated from heterozygous IL-23R-GFP+/− mice, were detected in the proximal duodenum of naive IL-23R GFP reporter mice. Consistent with previous reports, IL-23R expression was not found in epithelial cells but rather was localized to clusters of cells forming nodules within the LP, morphologically consistent with gut-associated lymphoid tissue (Figure 5a). In addition, IL-23-induced tumorigenesis could not be initiated in compound mutant mice deficient for both recombinase activating gene-2 (RAG2) and common cytokine receptor gamma chain (RAG2−/− × IL-2Rγc−/−) (Figure 5b), suggesting signaling through a lymphoid-lineage cell was required. These results indicate that the primary IL-23-responding cells are of immune origin and suggest that the dysregulated epithelial cell proliferation and dysplasia induced by IL-23 is a consequence of immune cell action.

Interleukin (IL)-17 and interferon-γ (IFNγ) have functional roles in IL-23-induced duodenal adenomas driven by immune cells. (a) Normal proximal duodenal tissue from IL-23 receptor green fluorescent protein (GFP) reporter mice was stained by hematoxylin and eosin and for GFP expression by immunohistochemistry for routine histological examination. Representative images are shown from at least five mice. Scale bars indicate 200 or 40 μm as shown. (b) Wild-type (WT) and RAG1−/− × IL-2Rγc−/− mice were injected with 5 μg of IL-23 minicircle DNA, and tumor development was quantified after approximately 8 weeks. Representative data from two independent experiments, with five mice per group are shown. (c) Gene expression analysis was performed as described in Figure 4. Averages and s.e.m. are shown (control n=5, non-tumor adj. n=24, tumor n=27). Statistically significant differences compared with control are indicated. (d) WT, RAG1−/−, and IL-17A−/− mice were injected with 3 μg of IL-23 minicircle DNA, and tumor development was quantified after approximately 150 days. Combined data from three independent experiments, with at least 11 total mice per group are shown. (e) WT and IFNγ−/− mice were injected with 3 μg of IL-23 minicircle DNA, and tumor development was quantified after approximately 60 days. Combined data from two independent experiments are shown; with at least 15 total mice per group. (f, g) WT and IL-17A−/− × RAG1−/− (f) and IL-22−/− (g) mice were injected with 5 μg of IL-23 minicircle DNA, and tumor development was quantified after 21 weeks (f) and 8 weeks (g), respectively. Representative data from two independent experiments are shown, with at least five mice per group. *P<0.05; **P<0.01; ***P<0.001. NS, not significant.
The results above implying a requirement for lymphoid lineage immune cell activation prompted us to investigate cytokine expression associated with tumorigenesis. Gene expression analysis of duodenal tissue revealed a trend for the elevated expression of IL-17 in IL-23-induced tumors, as well as IFNγ and IL-22 in both tumor and NTA tissue, compared with gut from control mice (Figure 5c). We assessed the effect of IL-23 expression in various cytokine and RAG1-deficient mice to further characterize which lymphoid cell populations and cytokine expression profiles were required for tumorigenesis.. IL-23 expression in IL-17-deficient mice exhibited a significantly reduced tumor incidence and burden compared with WT mice (Figure 5d). In addition, IFNγ was also determined to have a functional role in adenoma formation. The tumor load in IFNγ-deficient mice expressing IL-23 was significantly reduced, although the tumor incidence was similar to WT mice (Figure 5e). In contrast, IL-23-induced tumorigenesis in mutant mice deficient for IL-22 expression was equivalent to that observed in WT mice (Figure 5g).
Tumorigenesis in RAG1-deficient mice was examined to assess the relative requirement for lymphoid-lineage cells from adaptive lymphocyte vs. innate lymphoid populations responding to IL-23. Tumor formation in the RAG1−/− mice was the same as in WT C57BL/6 (Figure 5d). In addition, the relative inhibition of tumorigenesis in IL-17-deficient mice on the WT C57BL/6 and RAG1−/− background strains was equivalent, suggesting IL-17 expression in ILCs contributed significantly to IL-23-induced tumorigenesis (Figure 5f).
Both adaptive lymphocyte and ILC populations express Thy1 (CD90) and have previously been demonstrated to contribute to colitis.20 Therefore, we used antibody depletion using an anti-Thy1 monoclonal antibody (mAb) in WT C57BL/6 vs. RAG1-deficient mice to further assess the relative functional contribution of ILC populations for IL-23-induced duodenal tumors. Thy1+ cells were depleted by treatment with anti-Thy1 antibody following IL-23 MC HDD. Thy1 cell depletion was confirmed in the spleen and in the duodenal LP (Figure 6a) of representative mice from each treatment group. Thy1 cell depletion resulted in a significant reduction in tumor incidence and size in both WT mice and in RAG1-deficient mice (Figure 6c). Hence, in the absence of Th17 cells (i.e., in RAG1−/− mice), depletion of Thy-1+ ILCs substantially inhibited tumorigenesis, further illustrating the proficiency of IL-23 activation of ILC for de novo initiation of carcinogenesis and adenoma formation in the gut.

Thy1+ innate lymphoid cells drive interleukin (IL)-23-induced duodenal tumors. Wild-type or RAG1−/− mice were injected subcutaneously with 1 mg per dose of anti-Thy1 or rat immunoglobulin G2b (IgG2b) control antibody twice a week for 4 weeks. One day following the first dose of antibody, mice were injected with 2 μg IL-23 minicircle DNA via hydrodynamic delivery; mice were killed after 4 weeks. (a) Tumors were quantified, and splenocytes were isolated and stained for Thy1 and CD45 to determine the extent of Thy1 cell depletion. (b) Gut lamina propria (LP) cells from the proximal duodenum of three mice per group in a were isolated and stained for Thy1 and CD45 to determine the extent of Thy1 cell depletion. (c) Quantification of tumors from mice in a. *P<0.05; ***P<0.001. Representative data from two experiments shown, with at least eight mice per group. (d) Representative histogram of Thy1+CD45+ gut LP cell depletion.
IL-23 induces immune deviation of Thy1+ IL-23R+ lymphoid cells expressing IL-17 and IFNγ cytokines associated with tumorigenesis
IL-23-mediated immune deviation of lymphoid populations and cytokine expression profiles associated with initiation of tumorigenesis was assessed by flow cytometric analysis on proximal duodenum LP isolated from the region of tumor initiation. LP cells from IL-23R GFP reporter mice expressing IL-23 or control MC were analyzed to assess the effects of IL-23R signaling on relative proportional changes in lymphocyte and innate lymphoid populations and cytokine expression profiles (Figures 7 and 8). B220+ B lymphocyte and Nk1.1+ NK cell numbers were not modulated by IL-23 expression, whereas Thy1+IL-23R+ CD4+ T lymphocytes expanded more than 10-fold (2.8% in control vs. 36.8% of IL-23R+ cells in IL-23-stimulated LP cells; see Figure 7). Interestingly, the majority of Thy1+IL-23R+ LP cells were negative or had Low Sca1 expression in control mice, whereas, upon IL-23 expression, virtually all of the Thy1+IL-23R+ population of cells expressed Sca1, with the majority of these cells expressing high levels. Differential effects of IL-23 exposure on proximal duodenal LP subsets of Group 3 ILCs28 were observed (Figure 7b). Stimulation of IL-23R induced an eightfold expansion of IL-23R+ Thy1+Sca-1+ CD3− ILC populations in IL-23 MC-treated mice compared with controls while NCR+ ILC3 populations (IL-23R+ Thy1+NKp46+ CD3−) expanded about twofold. In contrast, IL-23 expression induced a threefold decrease in lymphoid tissue inducer (IL-23R+ Thy1 +cKIT+ NKp46− CD3−) cell numbers.

Interleukin (IL)-23 modulates adaptive and innate lymphoid populations within duodenal lamina propria (LP). IL-23 receptor green fluorescent protein (GFP) reporter mice were injected with control (albumin) or IL-23 minicircle DNA and killed at 2 weeks post injection. (a) Phenotypic analysis of Thy1+GFP+CD45+-gated cells isolated from the duodenal LP using specific antibodies (solid line) and isotype controls (dotted line). Representative data from at least two independent experiments are shown. (b) Representative cytometric analysis of IL-23R+ (GFP) T lymphocyte and group 3 innate lymphoid cell (ILC) populations within proximal duodenal LP. (c) Quantification of cells analyzed in b. Three independent experiments were performed. **P<0.01; ***P<0.001. LTi, lymphoid tissue inducer; NK, natural killer; TCRβ, T-cell receptor β.

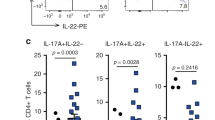
Interleukin (IL)-23 induced cytokine expression in duodenal lamina propria (LP) cells. IL-23 receptor green fluorescent protein reporter mice were injected with 5 μg control (albumin) or IL-23 minicircle (MC) DNA and killed 2-weeks post injection. Duodenal LP cells were isolated and pooled from three sets of four pooled mice for each treatment group. (a) Relative numbers of IL-23R+ LP cells in proximal duodenum were gated as shown in Figure 7b and shown as cells per gut. (b and c) Relative proportions of total proximal duodenal LP–derived CD3− innate lymphoid cells (ILC) with potential for expressing IL-17, IL-22, and interferon-γ (IFNγ were assessed by intracellular cytokine staining (ICS) and quantified as the percentage of total LP. (d) Relative proportions of CD3+CD4+ T-cells with potential for expressing IL-17, IL-22, and IFNγ were assessed by ICS and quantified as the percentage of total LP. Representative data from three independent experiments are shown. **P<0.01; ***P<0.001. SSC, side scatter.
Collectively, the results described above indicated that IL-23R signaling was sufficient to induce expansion of IL-23R+ cells from CD4+ T lymphocyte and Thy1+ CD3- ILC populations that have previously been associated with colitogenic properties in various IBD models.20 The relative numbers of IL-23R+, ILCs, and CD4+ T cells in the LP of the proximal duodenum tumor initiation region are shown in Figure 8a. We assessed the potential cytokine expression phenotypes of these respective populations by flow cytometry using phorbol myristate acetate (PMA) plus ionomycin stimulation and intracellular cytokine staining on total LP cell preparations (Figure 8b–d). Although the total numbers of ILCs increased in mice expressing IL-23, the relative proportion of capable of expressing IL-17 or IL-22 was not significantly changed (Figure 8c), whereas the proportion of ILCs capable of expressing IFNγ were significantly increased in IL-23 MC expressing mice. A similar trend was observed in CD4+ T-cell populations (Figure 8d). These data indicate that the expanded Thy1+IL-23R+ lymphoid cells in the duodenum are capable of producing key cytokines that were shown to have functional roles in IL-23-driven duodenal tumorigenesis (Figure 5).
IL-23 activates multiple cytokine responses associated with inflammation-induced tumorigenesis in the gut
IL-23 induces a pleiotropic array of inflammatory responses in the gut mediated in large part through induction of an increased expression cascade of numerous pro-inflammatory cytokines.29 Gene expression analysis of duodenal tissue was performed to identify IL-23-modulated cytokines that may contribute to tumorigenesis. Evidence for marked activation of pro-inflammatory molecular pathways, as illustrated by increased expression of IL-1β, IL-6, TNFα, and IL-11, were evident as early as 1–4 weeks (Figure 9a) and continued to be upregulated throughout tumor growth progression (Figure 9b). Expression of IL-1β, IL-6, IL-11, and TNFα mRNA was primarily elevated in the tumor and not in NTA tissue as compared with tissue from control mice. Hence, IL-23R stimulation was sufficient to induce tumor-specific expression of TNFα, IL-6, and IL-1β, previously shown to have roles in colorectal cancer (CRC) and cell growth, as well as IL-11, which has been shown to be involved in gastric cancer.1, 3, 4, 5, 6

Functional roles of cytokines in interleukin (IL)-23-driven adenoma development. Wild-type (WT) mice were injected with control or IL-23 minicircle and killed at (a) 1 or 4 weeks, (b–d) 21 weeks, or (f) 8–15 weeks post injection. (a, b) Gene expression analysis was performed as described in Figure 4. Averages and s.e.m. are shown (a: control n=5, non-tumor adjacent (NTA) n=24, tumor n=27, b: control n=3, 1 week n=5, 4 weeks n=5). Statistically significant differences compared with control are indicated. (c,d) WT, IL-6−/− (c), and tumor necrosis factor α−/− (TNFα−/−) (d) mice were injected with 3 μg of IL-23 minicircle DNA, and tumor development was quantified. Tumor data (c,d) is combined from three independent experiments with at least 11 total mice per group. (e) IL-23 receptor green fluorescent protein (GFP) reporter mice were injected with 2 μg control (albumin) or IL-23 minicircle DNA and killed 2 weeks post injection. Duodenal lamina propria cells were isolated and pooled from three mice and then stimulated with 50 ng ml−1 phorbol myristate acetate and 500 ng ml−1 ionomycin for 4 h. Cells were gated for GFP and Thy1. Data from two independent experiments shown. (f) WT mice were injected with 5 μg of IL-23, 6 μg of IL-17, 10 μg of IL-22 minicircle DNA individually, or 6 μg of IL-22 and 10 μg IL-17 minicircle DNA together in combination. Representative images of gross duodenal tissue from two independent experiments are shown with at least five mice per group in each experiment. Arrows indicate adenomas. The dotted line marks the pyloric sphincter. *P<0.05; **P<0.01; ***P<0.001.
To examine whether these cytokines identified above were in fact involved in IL-23-driven tumor development, IL-23 was expressed in mice deficient for IL-6 and TNFα. In IL-6−/− mice, adenoma growth was significantly reduced (Figure 9c), indicating that IL-6 had a functional role, as predicted from the mRNA expression data. In TNFα−/− mice, adenoma growth was enhanced and tumor load was significantly increased (Figure 9d). This result indicates that TNFα had a protective role in tumor growth. As IL-6 and TNFα were found to have functional roles in IL-23-driven duodenal tumors, protein expression of these cytokines was examined in Thy1+IL-23R+ pathogenic cells isolated from duodenal LP. Intracellular cytokine staining of LP cells stimulated with PMA/ionomycin revealed that TNFα was expressed in Thy1+IL-23R+ cells, whereas IL-6 was not (Figure 9e). Taken together, these data suggest that the Thy1+IL-23R+ lymphoid populations could also express TNFα and could induce pro-inflammatory IL-6 cytokine expression associated with initiation of tumorigenesis in other cell populations.
IL-23, but not IL-17 and IL-22, cytokine expression is sufficient for driving gut tumorigenesis
IL-23 mediates pro-inflammatory responses in the gut, in part, through the modulation of IL-23R+ Th17 and ILC, which in turn express IL-17 and IL-22.30, 31, 32 Cytokines IL-17 and IL-22 can directly induce receptor-positive gut epithelial cells to upregulate expression of antimicrobial proteins such as REG3β and S100A8, secretion of pro-inflammatory cytokines and chemokines (including CXCL1 (C-C motif chemokine ligand 1)), as well as increased proliferation and cell survival.3, 30, 31, 32 Indeed, the results shown in Figure 4 showed tumor-specific expression of IL-22, REG3β, S100A8, and CXCL1. We postulated that the IL-23-induced tumorigenesis may either be mediated through one of these nascent cytokine responses or may require an accumulative effect of multiple downstream regulatory effects. We utilized HDD of IL-17, IL-22, or a combination of both IL-17 plus IL-22 MC DNA vectors to evaluate these requirements. Only mice expressing IL-23 developed proximal duodenal tumors (Figure 9f), demonstrating a need for pleiotropic effects that could not be efficiently mediated by the expression of either IL-17 or IL-22 or a combination of both cytokines. These results underscore that IL-23 was both sufficient and required for induction of tumorigenesis in the gut.
Discussion
Recent genome-wide association studies of large IBD patient cohorts compared with healthy controls corroborate a fundamental contribution of IL-23 for IBD.9, 10 The fact that susceptibility for Crohn’s disease and ulcerative colitis were both linked to various single-nucleotide polymorphisms of the IL-23R gene confirmed the importance of signaling through IL-23R for IBD predisposition. The association of chronic inflammation and gastrointestinal cancer is exemplified in IBD patient populations. Although IBD-associated CRC represents only 2% of all CRCs, most of which are associated with other causes, the cumulative probability increases with chronic IBD duration.33 The incidence rate of CRC corresponded to cumulative probabilities of 2% by 10 years, 8% by 20 years, and 18% by 30 years of chronic intestinal inflammation.34 The importance of elucidating key inflammatory pathways that contribute to tumorigenesis is underscored by the effective use of non-steroidal anti-inflammory drugs, such as Cox-2 inhibitors, for the management of cancer risk in patients with Familial adenomatous polyposis.35, 36
The potential contribution of IL-23 as a tumor-promoting factor is emphasized by the observation that IL-23 expression is elevated in human CRC tumors.14 Moreover, induction of skin papillomas following application of a chemical carcinogen plus an inflammatory promoter (12-O-tetradecanoylphorbol-13-acetate) was inhibited in IL-23-deficient mice.14 IL-23 is clearly important for models of IBD and tumor development induced by pathogenic bacterial colonization, as neutralization with IL-23 or IL-23R-specific antibodies could inhibit disease.7, 8, 17 Nevertheless, there are too many factors in addition to IL-23/IL-23R associations with IBD and CRC to elucidate the specific roles IL-23 signaling may have in these complex pathological processes. The very complex array of inflammatory mediators and immune cell infiltrates render the capacity to investigate specific IL-23 contributions perplexing. This prompted us to develop a novel in vivo model to more effectively control for IL-23-specific regulation of cascading events culminating in tumor development in the absence of additional pathogenic, carcinogenic, or inflammatory stimuli.
We utilized HDD of MC DNA encoding an IL-23 transgene to induce exocrine expression from the liver.22, 23 No additional stimuli such as carcinogens, dextran sodium sulfate, anti-CD40 mAbs, pathogenic T cells, or pre-existing tumor-suppressor gene mutations such as APCmin were utilized. This provided us a relatively isolated and controlled perspective of IL-23-driven pathology and the capacity to investigate essential downstream regulatory pathways through the use of gene expression analysis, knockout mice, and antibody-depletion strategies. Interestingly, systemic exposure of IL-23 resulted in rapid onset of overt pathology in very specific sites, in which IL-23R-expressing sentinel cells reside. Namely, mice developed psoriatic-like lesions in the ears, arthropathies in the paws and spine,22, 23 and adenomatous tumors in the proximal duodenum with no other notable histopathology observed.
Remarkably, MC expression of IL-23 was sufficient to drive rapid de novo development of gut adenomas in the proximal duodenum of virtually all treated WT C57BL/6 mice. Neoplastic events occur very rapidly with clear evidence of proliferating cells advancing up the intestinal villi as early as 1 week following induction of IL-23 expression and development of macroscopic adenomas observed by 3 weeks. Notably, early tumorigenesis commenced in the absence of overt inflammatory infiltrates or relative increases in total LP cells, despite evidence of increased expression of biomarkers associated with other models of IBD, such as elevated S100A8, Reg3β, and MMP7. These results suggest IL-23R signaling, resulting in expansion and activation of a relatively small population of resident ILC, is sufficient to induce a potent set of tumor-initiating molecular pathways in mucosal villi epithelium. Additional inflammatory stimuli or the addition of carcinogens were not required. Moreover, normal commensal microbiota were sufficient as no infection with pathogenic bacteria such as H. hepaticus or Bacteroides fragilis expressing a pro-inflammatory enterotoxin was needed.17, 20, 37 To verify that Helicobacter strain infection was not required, fecal stool samples were analyzed at the IDEXX RADIL Reference Laboratories (Coumbia, MO) using a generic Helicobacter Spp. PCR assay that detects all bacteria in the Helicobacter genus and for specific strains for H. bilis, H. ganmani, H. hepaticus, H. rodentium, and H. typhlonius. Fecal samples from control C57BL/6 mice and tumor-bearing mice, expressing IL-23 MC for 4 or 16 weeks, all tested negative for Helicobacter colonization. A positive control strain, B6.IL-10 KO, were confirmed for the presence H. ganmani infection. In contrast to colon tumor induction with H. hepaticus plus the chemical carcinogen azoxymethane,35 IL-23-induced tumorigenesis was very efficient in both WT C57BL/6 and B6.RAG1−/− strains, which express a protective Hiccs locus (H. hepaticus–induced colitis) modulated in part by ILCs. Collectively, these results suggest that IL-23-induced molecular pathways are sufficient for initiation of gut tumorigenesis in the absence of exogenous carcinogens, Helicobacter colonization, and before development of overt inflammatory infiltrates.
Continued IL-23 expression was associated with progression of adenoma size, relative dysplasia, and development of extensive inflammatory infiltrates by 6 months. However, we did not observe any examples of malignant transformation resulting in invasion of the basement membrane, characteristic of progression to malignant carcinoma. The inflammatory infiltrates in more advanced adenomas were primarily limited to the tumor regions (data not shown). Tumors were limited in C57BL/6 mice to the proximal duodenum and were not observed in the colon. Importantly, while CRC is often associated with chronic IBD, a recent meta-analysis of IBD patients revealed an increased risk of cancer of the upper gastrointestinal tract with a standardized incidence ratio of 2.87.12
Tumor-specific upregulation of numerous pro-inflammatory cytokines reported to be contributing factors to tumor incidence and progression were observed as early as 1 week following initiation of IL-23 trangene expression. In particular, significant upregulation of IL-1β, IL-6, TNFα, and IL-11 expression was observed in IL-23-induced adenomas NTA compared with NTA tissue. Notably, tumor growth was significantly inhibited in mice deficient for IL-6 expression, a cytokine for which extensive data exist for gastrointestinal tumor development and progression.1, 3 On the other hand, tumor growth and progression was augmented in TNFα-deficient mice. Although TNFα has been associated with tumor progression, it has also been shown to be a critical cytokine for tumor immune surveillance.4, 38 Collectively, these data indicate that the IL-23-induced cascade of pro-inflammatory responses are sufficient to drive many of the pleiotropic cytokine expression profiles previously associated with inflammation-induced tumorigenesis and progression in the gut. The data also suggest that IL-23 may also induce immune tumor surveillance mechanisms regulated by TNFα as a counterbalance.
Important contributions of Th17 cells in promoting intestinal adenoma development have been shown in various APCMin models.16, 17 In addition, a potential role of Th17 cells in gastroduodenal pre-malignant polyp development in mice deficient for SMAD-4 signaling was recently implied owing to an increased expression of IL-1β, IL-6, TNFα and IL-11 and the presence of Th17 cell in the tumor lesions.18 It is important to note in our studies that IL-23 clearly induced expansion and activation of Th17 cells within the tumor initiation site in proximal duodenal LP. Moreover, tumorigenesis was inhibited in IL-17−/− mutant mice, suggesting Th17 cells may contribute to tumor initiation. However, we were not able to specifically assess the relative contribution of Th17 cells owing to the lack of experimental models in which ILCs are deficient while leaving Th17 populations intact.
We were, on the other hand, able to empirically evaluate the relative contribution of ILCs by utilizing RAG1-deficient mice in which IL-23 tumor initiation was equivalent to WT mice. Moreover, IL-23-induced tumorigenesis was inhibited in (RAG1−/− × IL-17−/−) double knockout mice providing evidence for an important contribution of IL-17 expression in ILC3s. Further support for the key role IL-23R+ ILCs play in tumorigenesis is illustrated in Th17-deficient RAG1−/− KO mice, in which anti-Thy1 mAb depletion of remaining ILC populations inhibited tumor development. These results provide the first direct evidence that group 3 ILCs are critical for IL-23 initiation of tumorigenesis, while the significant inhibition of tumorigenesis in IFNγ−/− mutant mice, as well as the expansion of ILCs expressing IFNγ, suggests a positive correlation. Directly assessing the requirement for IFNγ responses in IL-23R+ ILC will be addressed in future studies.
Although endogenous IL-17 contributed to tumor formation, th expression of IL-17, IL-22 or a combination of both cytokine MC transgenes was not sufficient to induce adenoma development. These results imply that neither the IL-17-induced downstream cytokines nor the IL-22 attributed cytoprotective and proliferative phenotypes were sufficient for initiating adenoma development, although IL-22 expression in ILC has recently been demonstrated to have a critical role for maintenance and progression of Helicobacter plus azoxymethane carcinogen–induced tumors in the colon.39 Collectively, these indicate that the IL-23-mediated responses required for tumor initiation are pleiotropic and that the essential initiating cells for these responses are ILCs expressing Thy-1, Sca-1, and IL-23R.
The capacity of chronic cytokine transgene expression to initiate tumorigenesis has been reported with notable differences from the IL-23-mediated tumorigenesis demonstrated here. Mice expressing an IL-6 transgene were shown to develop extraosseous plasmacytomas, from B-lymphocyte lineage cells, in about 50% of mice by 18 months of age.40 Overexpression of IL-1β transgenes initiates gastritis with inflammatory cell infiltrates resulting in gastric epithelium tumor development in about 70% of mice aged >1 year.24 In contrast, IL-23 transgene expression induces epithelial-derived adenomas in the proximal duodenum,within 3 weeks with virtually 100% incidence, before development of overt inflammation, and was dependent on a relatively small population of resident ILCs. In contrast to the effects of IL-23 on ILCs, paracrine expression of IL-12 has been shown to induce NKp46+ lymphoid tissue inducer group ILCs to suppress tumor growth in the B16 model for melanoma.41
Systemic IL-23 expression and the downstream regulatory pathways were sufficient for initiation of dysregulated epithelial cell proliferation in the proximal duodenum that leads to de novo adenoma formation, independent of carcinogens or Helicobacter colonization. These results support the premise that IL-23 is a key upstream regulatory cytokine, sufficient and essential for the induction of numerous cytokine responses associated with gut tumorigenesis. Moreover, induction of IL-23R expressing innate lymphoid sentinels resident in the LP of the upper gastrointestinal gut contribute to the nucleating events resulting in neoplastic transformation and tumorigenesis. These results suggest blocking IL-23 signaling in chronic IBD may have the potential of reducing relative cancer risk over time.
Methods
Mice. C57BL/6 mice were obtained from the Jackson Laboratory (Bar Harbor, ME). C57BL/6J IL-17−/− (B6.129-IL-17A(KO)), RAG1−/− (B6.129S7-Rag1 tm1Mom/J), IFNγ−/− (B6.129-Ifng tm1), IL-6−/− (B6.129S2-IL-6tmkopf/J), TNFα−/− (B6/Culas-TNFtm1Cent), IL-23R−/− (B6.129S6-IL-23RKO(neo-flipped)), IL-22−/− (C57BL/6-IL22<tm1 Xen>), RAG−/− × IL-2Rγc−/− (B6.129S6-Rag2 tm1Fwa yC tm1French), RAG−/− × IL-17A−/− (B6.129-Rag1<tm1Mom>IL-17A<tm1Kyo>/DNAX), and IL-23 GFP reporter (IL-23R (GFP/+) Het) were maintained under specific pathogen-free conditions at the animal facility of Merck Research Laboratories, Palo Alto, CA. All animal procedures were approved by the Institutional Animal Care and Use Committee of Merck Research Laboratories in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care.
MC DNA and HDD. MC DNA used for HDD was previously described in detail.23 HDD of IL-23 MC or control MC (hAAT or albumin) was performed according to the standard procedures; briefly, DNA amounts as indicated in respective figures were injected via the tail vein in a volume of sterile Ringer’s solution equivalent to 10% mouse body weight over 5–7 s. IL-23 expression was confirmed in serum for quality control verification of transgene expression efficiency between MC vector lots and from terminal cardiac bleeds using an IL-23 enzyme-linked immunosorbent assay.22
Tumor quantification. Tumors were photographed and were manually identified from the photographs using the Axio Vision software (Carl Zeiss MicroImaging GmbH, Jena, Germany). Tumor load/burden was calculated by the summation of all tumor areas, as determined by the software, per mouse.
RNA extraction. Total RNA was isolated from tissue by homogenization into RNA STAT-60 (Tel-Test, Friendswood, TX) using a polytron homogenizer and extracted according to the manufacturer’s instructions. After isopropanol precipitation, total RNA was re-extracted with phenol:chloroform:isoamyl alcohol (25:24:1; Sigma-Aldrich, St. Louis, MO) using phase-lock light tubes (5 Prime, Thermo Fisher Scientific, Pittsburgh, PA).
Real-time quantitative PCR (qRT-PCR). DNase-treated total RNA was reverse-transcribed using QuantiTect Reverse Transcription (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Primers were designed using Primer Express (Applied Biosystems, Life Technologies, Foster City, CA) or obtained commercially from Applied Biosystems. qRT-PCR on 10 ng of cDNA from each sample was performed using either of the two methods. In the first method, two gene-specific unlabelled primers were utilized at 400 nM in an Applied Biosystems SYBR green qRT-PCR assay utilizing an ABI 7300. In the second method, two unlabelled primers at 900 nM each were used with 250 nM of FAM-labeled probe (Applied Biosystems) in a TAQMAN qRT-PCR reaction on an ABI 7300. The absence of genomic DNA contamination was confirmed using primers that recognize genomic region of the CD4 promoter. Ubiquitin levels were measured in a separate reaction and were used to normalize the data by the Δ-Δ Ct method. (ABI User Bulletin no. 2, 1997) (The equation 1.8(Ct ubiquitin−Ct gene of interest) × 104 was used to obtain the normalized values.)
Histology and immunohistochemistry. Tissue sections were evaluated by two board-certified pathologists (JY and RP). Formalin-fixed paraffin-embedded blocks of duodenal tissue were sectioned at 4 μm, then deparaffinized, and rehydrated with serial passage through changes of xylene and graded ethanols for routine hematoxylin and eosin staining and IHC. For Ki67, CD3, F4/80, and MPO IHC, slides were subjected to antigen retrieval in target retrieval solution pH 6.1 buffer (S1699, Dako, Carpineteria, CA) at 120 °C for 4 min, followed by cooling to 90 °C for 10 min. Endogenous peroxidase in tissues was blocked by incubation of slides in 3% hydrogen peroxide solution before incubation with primary antibody (anti-Ki67 (clone TEC-3, Dako); anti-CD3 (clone CD3-12, AbD Serotec, Raleigh, NC); anti-F4/80 (clone BM8, eBioscience, San Diego, CA); or anti-MPO (rabbit polyclonal, Dako)) for 60 min. Anti-Ki67 primary antibody was detected via application of biotinylated secondary antibody (712-066-153, Jackson ImmunoResearch, West Grove, PA) and horseradish peroxidase–conjugated avidin (ABC Elite, PK7100, Vector Laboratories, Burlingame, CA). Anti-CD3 and anti-F4/80 primary antibodies were detected using Rat-on-Mouse HRP Polymer (RT517L, Biocare Medical, Concord, CA). Anti-MPO was detected using the EnVision+ System–HRP (K4011, Dako). Antigen–antibody binding was visualized with 3,3′diaminobenzidine chromogen (K4368, Dako). For GFP IHC, slides were stained using the CSA II Biotin-Free Tyramide Signal Amplification System (K1497, Dako) according to the manufacturer’s instructions. Slides were subjected to antigen retrieval with a 5-min Proteinase K incubation (S3020, Dako) before a 60-min incubation with anti-GFP antibody (A11122, Invitrogen, Grand Island, NY).
Thy1 depletion. Anti-Thy1 (30H12) or rat IgG2b (LTF-2) control antibodies (Bio X Cell, West Lebanon, NH) were injected subcutaneously at 1mg per mouse twice a week until experiment termination. The first dose of anti-Thy1 antibody was given 1 day before HDD of 2 μg IL-23 MC DNA. Splenocytes were prepared by standard procedures. Gut LP cells were isolated by first removing epithelial cells through the incubation of 0.5-cm gut tissue pieces in Hank’s buffered salt solution containing 5 mM EDTA and 10 mM HEPES for 20 min at 37 °C and then repeating this incubation one additional time. The remaining tissue was cut into small fragments and then digested with complete RPMI medium containing 2mg ml−1 Collagenase Type I (Worthington Biochemical, Lakewood, NJ) and 30 U ml−1 DNaseI (Sigma-Aldrich, St. Louis, MO) at the same conditions. The resulting cell suspension was layered on to a 40%/80% Percoll gradient and centrifuged for 30 min at 600 g; LP cells were recovered at the interface. Cells were stained with antibodies against Thy1 and CD45 (BD Biosciences, San Jose, CA), and data were acquired on a BD FACSCalibur and analyzed with FlowJo software (Tree Star, Ashland, OR).
Gut LP cell analysis. After gut LP cells were isolated as described above, cells were cultured in complete RPMI with GolgiPlug (BD Biosciences) and stimulated with 50 ng ml−1 PMA (Sigma) and 500 ng ml−1 ionomycin (Sigma) for 4 h. Cells were then stained for respective surface markers and intracellular cytokines using the Cytofix/Cytoperm kit from BD Biosciences. Data were acquired on a BD FACSCantoII and analyzed with FlowJo software (Tree Star). Antibodies against mouse CD45 (30-F11), CD90.2 (Thy1, 53-2.1), Ly6C/G (Gr1, RB6-8C5), Ly6A/E (Sca1, D7), B220 (RA3-6B2), CD11b (M1/70), CD117 (cKIT, 2B8), CD4 (RM4-5), NKp46 (29A1.4), CD44 (IM7), CD25 (PC61), CD127 (IL-7Rα, SB/199)), IL-17A (TC11-18H10), IL-6 (MP5-20F3), and TNFα (MP6-XT22) were purchased from BD Biosciences. Antibodies against mouse NK1.1 (PK136), IFNγ (XMG1.2), and IL-22 (1H8PWSR) were purchased from eBioscience (San Diego, CA).
Statistical analysis. Statistical analysis was performed using the GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). Data were analyzed with the Mann–Whitney or Kruskal–Wallis test where appropriate.
References
Terzic, J., Grivennikov, S., Karin, E. & Karin, M. Inflammation and colon cancer. Gastroenterology 138, 2101–2114 e5 (2010).
Grivennikov, S.I., Greten, F.R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Popivanova, B.K. et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570 (2008).
Ernst, M. et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. J. Clin. Invest. 118, 1727–1738 (2008).
Kaler, P., Augenlicht, L. & Klampfer, L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene 28, 3892–3902 (2009).
McKenzie, B.S., Kastelein, R.A. & Cua, D.J. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 27, 17–23 (2006).
Onishi, R.M. & Gaffen, S.L. Interleukin-17 and its target genes: mechanisms of interleukin-17 function in disease. Immunology 129, 311–321 (2010).
Duerr, R.H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006).
Consortium., W.T.C.C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Xie, J. & Itzkowitz, S.H. Cancer in inflammatory bowel disease. World J. Gastroenterol. 14, 378–389 (2008).
Pedersen, N. et al. Risk of extra-intestinal cancer in inflammatory bowel disease: meta-analysis of population-based cohort studies. Am. J. Gastroenterol. 105, 1480–1487 (2010).
Bernstein, C.N., Blanchard, J.F., Kliewer, E. & Wajda, A. Cancer risk in patients with inflammatory bowel disease: a population-based study. Cancer 91, 854–862 (2001).
Langowski, J.L. et al. IL-23 promotes tumour incidence and growth. Nature 442, 461–465 (2006).
Mumm, J.B. & Oft, M. Cytokine-based transformation of immune surveillance into tumor-promoting inflammation. Oncogene 27, 5913–5919 (2008).
Grivennikov, S.I. et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491, 254–258 (2012).
Wu, S. et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 15, 1016–1022 (2009).
Hahn, J.N., Falck, V.G. & Jirik, F.R. Smad4 deficiency in T cells leads to the Th17-associated development of premalignant gastroduodenal lesions in mice. J. Clin. Invest. 121, 4030–4042 (2011).
Geremia, A. et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 208, 1127–1133 (2011).
Buonocore, S. et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010).
Wiekowski, M.T. et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J. Immunol. 166, 7563–7570 (2001).
Sherlock, J.P. et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat(+) CD3(+)CD4(-)CD8(-) entheseal resident T cells. Nat. Med. 18, 1069–1076 (2012).
Adamopoulos, I.E. et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J. Immunol. 187, 951–959 (2011).
Tu, S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008).
Cox, J.H. et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 5, 99–109 (2012).
Parham, C. et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 168, 5699–5708 (2002).
Awasthi, A. et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J. Immunol. 182, 5904–5908 (2009).
Spits, H. et al. Innate lymphoid cells—a proposal for uniform nomenclature. Nat. Rev. Immunol. 13, 145–149 (2013).
Maloy, K.J. & Kullberg, M.C. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 1, 339–349 (2008).
Cua, D.J. & Tato, C.M. Innate IL-17-producing cells: the sentinels of the immune system. Nat. Rev. Immunol. 10, 479–489 (2010).
Brand, S. et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G827–G838 (2006).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Triantafillidis, J.K., Nasioulas, G. & Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 29, 2727–2737 (2009).
Munkholm, P. Review article: the incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 18 (Suppl 2), 1–5 (2003).
Huls, G., Koornstra, J.J. & Kleibeuker, J.H. Non-steroidal anti-inflammatory drugs and molecular carcinogenesis of colorectal carcinomas. Lancet 362, 230–232 (2003).
Baron, J.A. & Sandler, R.S. Nonsteroidal anti-inflammatory drugs and cancer prevention. Annu. Rev. Med. 51, 511–523 (2000).
Boulard, O., Kirchberger, S., Royston, D.J., Maloy, K.J. & Powrie, F.M. Identification of a genetic locus controlling bacteria-driven colitis and associated cancer through effects on innate inflammation. J. Exp. Med. 209, 1309–1324 (2012).
Calzascia, T. et al. TNF-alpha is critical for antitumor but not antiviral T cell immunity in mice. J. Clin. Invest. 117, 3833–3845 (2007).
Kirchberger, S. et al. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 210, 917–931 (2013).
Kovalchuk, A.L. et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc. Natl. Acad. Sci. USA 99, 1509–1514 (2002).
Eisenring, M., vom Berg, J., Kristiansen, G., Saller, E. & Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 11, 1030–1038 (2010).
Acknowledgements
These studies were performed at and supported by Merck Research Laboratories.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
All the authors were former or are current employees of MRL. The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Chan, I., Jain, R., Tessmer, M. et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol 7, 842–856 (2014). https://doi.org/10.1038/mi.2013.101
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2013.101
This article is cited by
-
Innate lymphoid cells and innate-like T cells in cancer — at the crossroads of innate and adaptive immunity
Nature Reviews Cancer (2023)
-
The unique role of innate lymphoid cells in cancer and the hepatic microenvironment
Cellular & Molecular Immunology (2022)
-
Plasticity of innate lymphoid cell subsets
Nature Reviews Immunology (2020)
-
Interleukin 23 and autoimmune diseases: current and possible future therapies
Inflammation Research (2020)
-
Colorectal carcinoma in the course of inflammatory bowel diseases
Hereditary Cancer in Clinical Practice (2019)
