
Abstract
The discovery of the Th1/Th2 paradigm of CD4+ T-cell subsets redefined our understanding of immunity by highlighting the essential roles of cytokine networks in the induction and regulation of immune responses. Most recently, the identification of an additional subset, known as Th17 cells, has further illustrated the complexity and diversity of effector CD4+ T cells. Th17 responses have been closely associated with the cytokine interleukin (IL)-23 and, although originally pinpointed as having a deleterious role in autoimmune tissue pathology, the IL-23/Th17 axis has also been associated with protective immunity at mucosal surfaces. Recent progress has highlighted the heterogeneous nature of Th17 responses, has demonstrated diverse cellular sources for Th17-associated cytokines, and has begun to dissect the individual roles of these cytokines in different disease processes. Here, we will review the evidence linking the IL-23/Th17 axis to chronic intestinal inflammation and also will discuss its beneficial roles in intestinal protection and homeostasis.
Similar content being viewed by others


Introduction
Human inflammatory bowel diseases (IBDs) are chronic dis-orders of the gastrointestinal tract associated with aberrant activation of host immune responses toward constituents of the host intestinal microflora. 1, 2 The incidence of IBD is around 0.2% in developed countries and with no currently available cure, life-long immune-suppressive therapy and surgical intervention may be required.3 IBD encompasses a heterogeneous group of diseases with two main forms, Crohn's disease (CD) and ulcerative colitis (UC), which are distinguished by their clinical and pathological features.3 Although the precise etiology of IBD is uncertain, our understanding of these complex disorders has been greatly aided by the development of a variety of murine models that have provided tractable systems with which to study IBD pathogenesis. Although these models are themselves highly heterogeneous, with different models replicating different aspects of the human disease, they have confirmed the multi-factorial nature of IBD and highlighted how defects in epithelial barrier function or in host innate or adaptive immune responses can precipitate IBD pathology.1, 2 The murine IBD models have also emphasized that diverse triggering stimuli can converge on common downstream immune effector pathways. In parallel, increasingly powerful genome-wide analysis techniques have facilitated progress in identifying host genetic susceptibi-lity loci in human IBD patients. The major current challenge is to reconcile these disparate data sets and to relate the human genetic susceptibility data into potential functional effects that can subsequently be assayed using appropriate murine models. Of particular interest is the identification of cytokine networks that play a dominant role in either induction or regulation of chronic intestinal inflammation as these open up new opportunities for therapeutic intervention.4
Identification of a Key Role for IL-23 in IBD
Interleukin (IL)-23 is a heterodimeric cytokine belonging to the IL-12 family. While IL-12 is composed of an IL-12p40 subunit complexed with an IL-12p35 subunit, the closely related IL-23 comprises the IL-12p40 subunit complexed with the unique IL-23p19 subunit.5, 6 As IL-23 was discovered only relatively recently, early studies that associated IL-12p40 with many chronic inflammatory disorders, including IBD, were interpreted as reflecting a dominant role for IL-12-driven Th1 responses in disease pathogenesis.7 In the case of IBD, this hypothesis was strengthened by findings in animal models that genetic ablation or antibody-mediated depletion of IL-12p40 blocked intestinal inflammation.8, 9, 10, 11 and 12 Similarly, intestinal lesions in human CD patients exhibited elevated levels of IL-12 and associated Th1 cytokines.13, 14 These findings culminated in clinical trials that reported beneficial effects following administration of anti-IL-12p40 antibodies to patients with active CD.15, 16 However, the discovery of IL-23 and the realization that anti-IL-12p40 antibodies would neutralize both IL-12 and IL-23 prompted a re-evaluation of their relative roles in chronic inflammatory diseases.
Pioneering studies in murine models of autoimmune diseases, such as encephalomyelitis and collagen-induced arthritis, demonstrated that IL-23 played an essential role in driving autoimmune tissue pathology17, 18 and that this was associated with the accumulation of a novel subset of inflammatory CD4+ T cells secreting IL-17A, subsequently termed Th17 cells.19, 20, 21 and 22 Convincing evidence that this novel IL-23/Th17 axis played a key role in chronic intestinal inflammation was provided by parallel investigations in several different murine IBD models. Targeted depletion of IL-23, either by genetic ablation of the IL-23p19 gene or by using monoclonal antibodies specific for the IL-23p19 subunit, caused a marked attenuation of T cell-dependent colitis in T-cell transfer models of IBD23, 24 and also inhibited spontaneous colitis development in IL-10−/− mice.25 Furthermore, IL-23 was also required for the induction of intestinal inflammation by innate immune activation, as neutralization of IL-23 blocked the development of typhlitis and colitis in RAG−/− mice that was induced either by infection with the enteric pathogen Helicobacter hepaticus23 or by administration of agonistic anti-CD40 antibodies.26 In contrast, selective depletion of IL-12 had no effect on the development of innate immune-mediated or T cell-dependent intestinal inflammation.23, 24, 25 and 26 A second important paradigm to emerge from these studies was that colitis was associated with a marked elevation of IL-23 in the inflamed intestine, but not in systemic tissues such as the liver and spleen.23, 26 Moreover, systemic immune pathology was not abrogated in the absence of IL-23 but was instead dependent on IL-12.23, 26 This suggests that IL-23 and IL-12 may have divergent roles in mucosal and systemic immune responses and that selective depletion of IL-23 might abrogate intestinal inflammation while sparing systemic immune responses. The therapeutic potential of IL-23 targeting was recently confirmed in a study showing that treatment with anti-IL-23p19 antibodies ameliorated established colitis induced by adoptive transfer of an enteric bacteria-reactive CD4+ T-cell line.27 Similarly, the beneficial effects of anti-IL-12p40 antibody treatment in human CD patients were associated with a reduction in IL-23 secretion by lamina propria (LP) mononuclear cells.16 Together, these findings highlight a crucial role for IL-23 in chronic intestinal inflammation and validate the IL-23 axis as a potential therapeutic target.
Importantly, recent comprehensive genome-wide association studies of large cohorts of IBD patients and healthy controls have confirmed a central role for IL-23 in human IBD. These extensive studies identified several single-nucleotide polymorphisms in the IL-23R gene locus that were associated with either susceptibility or resistance to IBD.28, 29 A number of additional genetic studies replicated the association of IL-23R gene polymorphisms with CD and also confirmed associations with UC, indicating that genetic variants in IL-23R may contribute to both UC and CD susceptibility.28, 29, 30, 31, 32 and 33 It is not yet clear how IL-23R polymorphisms might predispose to IBD, but the identification of both disease-protective and risk-associated IL-23R variants28, 29 suggests that delicate regulation of IL-23R signals may play a crucial role in maintaining immune homeostasis in the intestine. The major protective IL-23R variant allele results in an amino-acid change (Arg381Gln) in the cytoplasmic domain of the IL-23R, suggesting that its protective effect may be due to disruption of IL-23R signaling.28 It has been speculated that the IBD-associated non-coding single-nucleotide polymorphisms found in IL-23R intronic regions might influence the production of different splice isoforms of the IL-23R.30, 34 and 35 IL-23R signals through the Signal Transducer and Activator of Transcription-3 (STAT3) pathway,6 and it is notable that genome-wide association studies also identified a single-nucleotide polymorphism within the STAT3 locus as being linked to CD.29 The IL-23R is not expressed on naive T cells, but is predominantly expressed by activated T cells and natural killer (NK) cells, as well as at lower levels by monocytes, macrophages, and dendritic cell (DC) populations.6 Therefore, polymorphisms affecting IL-23R signaling may potentially influence both innate and adaptive immune responses, and identifying the consequences of IL-23R polymorphisms on the functions of different leukocytes represents an important future goal. The difficulty of this challenge should not be underestimated; the fact that 7 years of intensive investigation has failed to yield any consensus on how mutations in the CARD15/NOD2 innate immune receptor predispose to the development of CD should serve as a cautionary tale.36, 37 Insights into the role of IL-23R signals in IBD pathogenesis might be facilitated by development and analysis of mice harboring cell type-specific deletions of the IL-23R. Improved understanding of IL-23R signaling may have benefits beyond IBD, as IL-23R polymorphisms have also been associated with other chronic inflammatory diseases, including ankylosing spondylitis38 and psoriasis,39 where anti-IL-12p40 treatment was successfully employed in a recent clinical trial.40 In addition, IL-23 is a potent tumor-promoting factor and is elevated in human colorectal tumors,41 suggesting that therapies targeting IL-23 may represent potential prophylactic treatments for inflammation-associated colon cancer in IBD patients.
Like other members of the IL-12 family, IL-23 appears to be secreted by DC primarily in response to microbial stimulation,6 but the factors that determine whether an activated DC will preferentially produce IL-12 or IL-23 remain poorly understood. However, emerging evidence suggests that distinct combinations of pattern-recognition receptors and their associated signal transduction pathways may trigger differential responses.42 Although many TLR agonists appear to stimulate synthesis of both IL-12 and IL-23, commensal Gram-negative bacteria or peptidoglycan activation of TLR2/NOD2 pathways favored IL-23 production43, 44, 45 and 46 as did triggering of the dectin family of C-type lectin receptors47, 48 or activation of purinergic receptors by extracellular ATP.49 Preferential stimulation of IL-23 production by peptidoglycan and ATP raises the possibility that activation of specific cytosolic NOD-like receptors (NLR) may contribute to the induction of IL-23.50, 51 Additional evidence for a role for the intestinal microflora in induction of IL-23 was provided in an elegant study of transgenic mice expressing a luciferase reporter gene under the control of the IL-12p40 promoter.52 It was found that under normal physiological conditions, IL-12p40 expression was restricted to a population of lamina propria CD11b− DC in the terminal ileum, a site that is often affected in patients with CD, and that this IL-12p40 expression was predominantly associated with IL-23p19.52 In addition, many of these LPDCs contained intracellular bacteria, and IL-23-expressing LPDCs were not detectable in germ-free mice.52 In this respect, it is also worth noting that although Th17 cells are relatively scarce in peripheral lymphoid tissues, they are significantly more abundant in the intestinal LP.53, 54 In fact, it was recently found that a population of CD11b+ LPDC isolated from the small intestine preferentially induced the differentiation of Th17 cells in a transforming growth factor-β (TGF-β)-dependent manner.55 Taken together, these findings support the hypothesis that the intestine is a site that is particularly conducive to IL-23/Th17 responses and that LPDC may facilitate the induction and maintenance of Th17 responses in the gut. Finally, although microbial signals may be the most abundant stimuli for intestinal IL-23 production, the induction of IL-23-dependent colitis by administration of agonistic anti-CD40 mAb indicates that costimulatory signals derived from activated T cells can potentiate IL-23 production in the intestinal mucosa.26
IL-23 Orchestrates both Pathogenic and Protective Immune Responses in the Gut
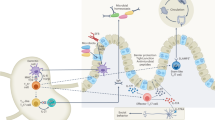
In terms of the molecular mechanisms by which IL-23 promotes colitis, current evidence suggests that IL-23 drives chronic intestinal inflammation through the induction of a diverse range of inflammatory responses. T cell-mediated colitis was associated with increased expression of a plethora of pro-inflammatory cytokines and chemokines in the gut, including IL-1β, keratinocyte-derived cytokine, monocyte chemoattractant protein-1, IL-6, tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ), and IL-17A, which were markedly attenuated in the absence of IL-23.23, 24 and 27 Analyses of mucosal CD4+ T-cell responses in two of these studies indicated that IL-23-dependent T cell-mediated colitis was associated with strong Th1 responses, with a minor population of Th17 cells also present.23, 24 However, anti-IL-23p19 antibodies also ameliorated the colitis that arose following adoptive transfer of a bacteria-reactive CD4+ Th17 cell line, a finding associated with increased apoptosis of the Th17 cells.27 Thus, IL-23 appears to promote the expression of pathogenic Th1 and Th17 responses in the intestine. Somewhat surprisingly, despite the complete absence of T cells, the innate immune typhlocolitis triggered by H. hepaticus infection of 129RAG−/− mice was characterized by an almost identical pattern of pro-inflammatory cytokines and chemokines as found in T cell-mediated IBD, which was again attenuated by treatment with anti-IL-23p19 antibodies.23 This innate immune pathology was characterized by an accumulation of granulocytes and monocytes in the inflamed intestine, and both populations expressed high levels of IL-17A.23, 56 IL-23 can trigger the release of pro-inflammatory cytokines such as IL-1β, IL-6, and TNF-α from myeloid cells,6 which in turn stimulates secretion of additional pro-inflammatory mediators by stromal, endothelial, and epithelial cells. Taken together, these observations indicate that IL-23 triggers a diverse pro-inflammatory cascade in the gut, comprising both acute innate responses as well as pathogenic adaptive responses, which, if left unchecked, can lead to the development of chronic intestinal inflammation (Figure 1a).

Protective and pathogenic roles of IL-23 and Th17 cytokines in the large intestine. (a) Aberrant immune regulation or pathogen encounter may activate DC to produce high amounts of IL-23, which will further activate innate immune cells such as DCs and monocytes to secrete pro-inflammatory cytokines like IL-1β, TNF-α, and IL-6. IL-23 also drives increased production of IFN-γ and Th17 cytokines by T cells and non-T cells, as well as impeding Treg cell activity. In this inflammatory environment, the major action of Th17 cytokines may be to promote additional inflammatory cascades by stimulating the production of chemokines that recruit and activate granulocytes and by triggering the release of tissue-degrading MMPs. Positive-feedback loops, such as CD40L–CD40 signals from activated T cells, may lead to sustained IL-23 production and further exacerbate chronic intestinal pathology. (b) When the inflammatory stimulus is removed, or when Treg cell activity becomes dominant, immune-suppressive cytokines like IL-10 and TGF-β act to resolve the inflammation. During this phase, Treg cell-induced TGF-β may act together with residual inflammatory cytokines to maintain Th17 activity. During resolution, Th17-associated cytokines may aid restitution and repair of the epithelial barrier while also fortifying barrier defences that inhibit bacterial colonization. DC, dendritic cell; IFN-γ, interferon-γ; IL, interleukin; MMPs, matrix metalloproteinases; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor-α.
A recent study revealed that an additional means through which IL-23 may exacerbate chronic intestinal inflammation is by constraining Treg activity in the intestine.57 Thus, in the T-cell transfer model of colitis, naive CD4+ T cells were able to induce moderate intestinal inflammation in IL-23p19−/−RAG−/− recipients when immune-suppressive cytokines were concomitantly neutralized.57 Moreover, naive CD4+ cells from Foxp3−/− mice induced severe colitis in IL-23p19−/−RAG−/− recipients, demonstrating that IL-23 was dispensable for colitis induction in the absence of Foxp3+ Treg activity. Although a previous study suggested that IL-23 may directly inhibit FoxP3 expression by T cells,58 IL-23 did not inhibit TGF-β-mediated FoxP3 induction in naive T cells in vitro,57 so it is not yet clear whether IL-23 acts directly or indirectly via other factors to override Treg activity in the gut. Nevertheless, these results emphasize that IL-23 may control the balance between effector and regulatory T cells in the intestine and also that, in situations where Treg activity is perturbed, colitis may arise through IL-23-independent pathways.
It is important to note that although ablation of IL-23 highly attenuated both innate and T cell-mediated forms of colitis, some residual inflammation persisted, which was not the case when IL-12p40 was depleted.23, 24 Further evidence that IL-12 can drive intestinal inflammation in the absence of IL-23 was reported in a study of 2,4,6-trinitrobenzene sulfonic acid-induced colitis, where IL-23p19−/− mice developed more severe intestinal pathology than wild-type (WT) mice, a response that was blocked by treatment with anti-IL-12p40 antibodies.59 These findings are consistent with previous studies that highlighted pathogenic roles for IL-12 and IFN-γ in intestinal inflammation.8, 9, 10, 11, 60 and 61 However, the precise relationship between IL-23 and IL-12 responses in the intestine remains to be elucidated, and they exhibit an important overlapping ability to promote pathogenic Th1 responses. Thus, both IL-12-dependent and IL-23-dependent mechanisms can contribute to intestinal pathology and may even act synergistically to induce maximal inflammatory responses.
Although most studies thus far have focused on the deleterious effects of IL-23/Th17 responses in host tissue pathology, evidence is emerging that this axis is also involved in mucosal immune defense.62 This host-protective role was first described in models of pulmonary infection with Klebsiella pneumoniae, where IL-23p19−/− and IL-17RA−/− mice exhibited greatly increased susceptibility and mortality.63, 64 Subsequent studies have implicated the role of IL-23/Th17 axis in protection from a number of pulmonary infections, including Mycoplasma pneumoniae,65Mycobacterium tuberculosis,66 and Bordetella pertussis.67 Furthermore, IL-23p19−/− mice also exhibited enhanced susceptibility and mortality following infection with the attaching and effacing (A/E) intestinal bacterium Citrobacter rodentium,54 which is a murine model of enteropathogenic Escherichia coli infection. In summary, because of its ability to orchestrate a range of innate and adaptive immune responses, IL-23 is an essential component of the host-protective response at mucosal surfaces, particularly against extracellular bacterial infections. Furthermore, as will be discussed more fully below, there is a growing appreciation that the IL-23/Th17 axis also regulates restitution and repair responses in the intestinal epithelium. However, in circumstances where high levels of IL-23 are sustained for an extended period, the associated chronic inflammatory responses may be a major driver of IBD pathogenesis.
Development of Th17 Cells and Production of Th17-Associated Cytokines
As noted above, IL-23 was initially implicated in the differentiation of a new subset of CD4+ T lymphocytes called Th17 cells based on their production of IL-17A (reviewed in refs. 22,68). However, subsequent findings clearly demonstrated that IL-23 was not required for Th17 differentiation—in fact, the IL-23R is not expressed on naive T cells5, 19—but that activation in the presence of TGF-β plus IL-6 directed the development of this subset.54, 69 and 70 Parallel studies established that IL-17-producing CD4+ T cells differentiated through the actions of the specific transcription factor RAR-related orphan receptor-γt53 and represented an effector subset that was distinct from the Th1 and Th2 lineages.71, 72 The role of IL-23 in Th17 cell biology is still somewhat unclear, although it is believed that this cytokine acts on activated CD4+ T cells that have upregulated their IL-23R following T-cell receptor (TCR) stimulation in the presence of TGF-β plus IL-6 or IL-21.58, 73 and 74 Thus, IL-23 may act as an expansion/maintenance factor for Th17 cells.22, 68
A rapidly expanding body of literature has highlighted that Th17 cells represent a heterogeneous population, capable of secreting a diverse range of cytokines including IL-6, TNF-α, IFN-γ, IL-17A, IL-17F, IL-21, and IL-22.22, 68 In addition, human Th17 cells may also produce IL-26 and CCL20.75 Although little information exists on what factors determine the cytokine secretion profile of Th17 cells, the local cytokine microenvironment is likely to provide key cues. For example, recent in vitro findings suggest that primary stimulation in the presence of TGF-β plus IL-6 followed by subsequent activation in the presence of IL-23 drives an IL-17/IL-22-secreting population, whereas if IL-23 is omitted in secondary stimulations, an IL-17/IL-10 double-producing population develops.76
The strong association of IL-23 with chronic intestinal inflammation23, 24, 25 and 26 has directed attention toward a potential role of Th17 cells in IBD pathogenesis. In the remainder of this review, we will discuss both the pro-inflammatory and the tissue-protective roles of Th17 cells and their associated cytokines in the gut. Importantly, we want to emphasize that additional non-T-cell sources exist for these factors (Table 1) and that IL-23 may drive Th17 cytokine production from non-CD4+ T cells. Thus, rather than exclusively discussing Th17 cells in colitis/tissue repair, we will treat these effector molecules as “Th17-associated cytokines”, regardless of their cellular origin.
Tissue-Protective and Pro-Inflammatory Roles of IL-17A and IL-17F in the Intestine
Increased expression of IL-17 family members (IL-17A and/or IL-17F) has been reported in both human IBD77, 78 and animal models of the disease.23, 24 In addition, it has recently been reported that polymorphisms in IL-17A and IL-17F genes are associated with increased susceptibility to UC.79 As mentioned above, CD4+ T cells are an important source of IL-17A and IL-17F, and cells with a Th17 phenotype have indeed been isolated from CD biopsies,80 suggesting a possible link to patho-logy. Moreover, data from innate immune-mediated IBD models reported IL-17A expression by several non-T-cell populations in the gut,23 and immunohistological analysis of biopsy tissue from human IBD patients revealed that IL-17A was clearly produced by both CD3+ T cells and CD68+ monocyte/macrophage populations in the inflamed intestine.77 However, the functional significance of IL-17A and IL-17F in disease pathogenesis in humans remains to be determined.
Binding of IL-17A and/or IL-17F to receptors on myeloid and endothelial cells has been shown to promote the expression of IL-1β, IL-6, and TNF-α, and chemokines (e.g., IL-8) that are involved in neutrophil mobilization, and thus in driving chronic inflammation in the central nervous system and the lung.17, 81 On the basis of these findings, it was initially thought that IL-17A and/or IL-17F would also have pathogenic functions in the gut. The finding that neutralization of IL-17A exacerbated disease in the dextran sulfate sodium (DSS) colitis model was therefore somewhat of a surprise, as it suggested a tissue-protective role for this cytokine in the gut.82 This result could potentially be explained by the ability of IL-17A to stimulate the expression of claudins in intestinal epithelial cells, thereby mediating tight-junction formation and fortifying the mucosal barrier.83 Moreover, IL-17A has also been described to have anti-inflammatory functions by blocking TNF-α-induced RANTES expression in human colonic subepithelial myofibroblasts.84 Finally, although not yet described for gut epithelial cells, IL-17A has also been shown to induce mucin production (Muc5) and β-defensin-2 in epithelial cells of the airways.85 A recent report has provided further support for a disease-protective role for IL-17A in intestinal pathology. Thus, in comparison to WT controls, DSS-treated IL-17A−/− mice displayed increased weight loss and more severe diarrhea, as well as increased leukocyte infiltration and more severe disruption of the epithelium.86 In contrast, IL-17F−/− mice developed reduced intestinal inflammation following DSS treatment and exhibited milder disease symptoms than WT controls.86 Together, these studies indicate that IL-17A plays a disease-protective role in the intestinal tract, whereas IL-17F may exacerbate the disease. In a separate study, mice with an epithelial cell-specific deficiency in Act1, an adaptor protein involved in IL-17R signaling, also showed reduced severity of DSS-induced colitis, with less neutro-phil infiltration and reduced chemokine secretion.87 Taken together, these findings imply that during the acute intestinal inflammation induced by administration of DSS, the stimulation of IL-17F-driven pro-inflammatory responses predominates over the tissue-protective effects of IL-17A. Similar findings were observed in the 2,4,6-trinitrobenzene sulfonic acid colitis model as IL-17R−/− mice showed reduced colitis scores compared to WT controls, and overexpression of an IL-17R-IgG1 fusion protein resulted in attenuated colonic inflammation.88
While the above studies focused on chemically induced acute colitis models, the role of IL-17A has also been examined in the well-established T-cell transfer model of chronic intestinal inflammation induced by transfer of naive CD4+ T cells from WT mice into lymphopenic recipients.89, 90 Using a modified version of this model, in which colitis was induced by transfer of IL-10-deficient CD4+ T cells into RAG−/− recipients, it was found that administration of recombinant IL-23 accelerated the onset of colitis.25 Additional experiments demonstrated that while neutralization of either IL-6 or IL-17A alone did not significantly reduce the development of colitis in this model, combined treatment with both IL-6- and IL-17A-neutralizing antibodies significantly attenuated the disease, although intestinal inflammation was not completely inhibited.25 These data argued for the involvement of IL-6 and IL-17A in T cell-mediated colitis, but at the same time indicate that additional factors are required for maximal inflammation. Subsequent studies in T-cell transfer IBD models have examined the role of T cell-derived IL-17A in colitis pathogenesis. First, when intestinal inflammation was induced in severe combined immunodeficiency animals by transfer of cecal bacterial antigen-specific Th1 or Th17 CD4+ T-cell lines, it was found that on a per-cell basis, the Th17 cell line was more colitogenic than the Th1 cell line.27 Thus, adoptive transfer of as few as 104 cecal bacterial antigen-specific Th17 cells induced severe colitis, whereas injection of even 106 cecal bacterial antigen-reactive Th1 cells triggered only mild intestinal inflammation.27 However, the mechanism by which the cecal bacterial antigen-specific Th17 cell line induced colitis was not analyzed further, so it is unclear whether IL-17A and/or other Th17-associated cytokines were responsible for their enhanced colitogenic potential. Recently, the requirement for T cell-derived IL-17A in colitis induction was definitively assessed in two independent studies using the classic T-cell transfer model of IBD. These studies clearly demonstrated that T cell-derived IL-17A was not required for intestinal inflammation, as RAG−/− recipients of naive IL-17A−/− CD4+ T cells developed colitis indistinguishable from that observed in mice given naive WT CD4+ T cells.57, 91
In summary, these results indicate that IL-17A is dispensable for DSS-induced inflammation and that T cell-derived IL-17A is not required for T cell-dependent colitis. Although a role for non-T-cell sources of IL-17A in the latter model cannot be ruled out until adoptive transfer experiments have been performed using IL-17A−/−RAG−/− recipients, the results from the DSS and 2,4,6-trinitrobenzene sulfonic acid colitis models clearly indicate that IL-17A has tissue-protective functions during acute intestinal inflammation. In contrast, IL-17F appears to play a more pathogenic role in the intestinal tract, as mice deficient in this cytokine showed reduced susceptibility to DSS colitis. However, further experiments in the T-cell transfer model are required to define the potential roles of T cell-derived vs. non-T-cell-derived IL-17F in chronic intestinal inflammation.
IL-21 Exerts Pro-Inflammatory Effects in the Gut
A role for IL-21 in gastrointestinal inflammation was suggested by studies demonstrating enhanced levels of this molecule in tissue biopsies from patients with IBD, Helicobacter pylori infection, or celiac disease.92, 93 and 94 In the case of celiac disease, genetic studies have further identified risk variants in the region harboring the IL21 gene.95 IL-21 has also been implicated in several other human diseases, including systemic lupus erythematosus, multiple sclerosis, and type I diabetes, and data from experimental models of these disorders support a pathogenic role for IL-21.96, 97 For example, administration of IL-21 prior to the induction of encephalomyelitis enhanced clinical disease,98 and IL-21 blockade using an IL-21/Fc fusion protein was shown to ameliorate symptoms in a mouse model of systemic lupus erythematosus.99 Similarly, treatment with an IL-21R/Fc fusion protein was recently shown to attenuate gut inflammation in the DSS colitis model, and IL-21−/− mice were reported to be largely resistant to both DSS- and 2,4,6-trinitrobenzene sulfonic acid-induced colitis,100 indicating pro-inflammatory effects of IL-21 in the intestine.
IL-21 is mainly produced by activated CD4+ T cells,58, 73, 74 and 101 but NKT cells102, 103 and T follicular helper cells104, 105 can also secrete this cytokine. Among CD4+ T cells, the Th17 subset is the main producer of IL-21, which is thought to act in an autocrine loop, sometimes in combination with TGF-β, to further induce Th17 cells.58, 73 and 74 Importantly, both IL-21−/− and IL-21R−/− mice are defective in generating Th17 responses,58, 73 and 74 arguing for an essential role for this molecule in Th17 differentiation. This finding may be explained by the fact that IL-21 mediates enhanced expression of RAR-related orphan receptor-γt and IL-23R.58, 74
IL-21 acts on a variety of cells of the immune system, including B cells, CD4+ and CD8+ T cells, NK cells, and DCs.96, 97 In addition—and of potential importance for intestinal inflammation—the IL-21R is constitutively expressed on intestinal fibroblasts and gut epithelial cells.106, 107 Stimulation of intestinal fibroblasts with IL-21 led to enhanced secretion of matrix metalloproteinases, molecules that mediate mucosal tissue degradation.107 Moreover, stimulation of a colonic epithelial cell line with IL-21 resulted in enhanced synthesis of the T-cell chemoattractant MIP-3α/CCL20,106 and in vivo blockade of this chemokine attenuated lymphocyte recruitment into the intestine in DSS-induced colitis.108 Another mechanism by which IL-21 may augment or sustain pro-inflammatory responses in the intestinal tract is by enhancing IFN-γ production by T cells and NK cells.109, 110 Indeed, IL-21 has been reported to promote Th1 responses in biopsies from CD and celiac disease patients.92, 94 However, under other circumstances, IL-21 has been reported to have an inhibitory effect on IFN-γ expression and to promote Th2 responses,111, 112 and 113 suggesting that its effects on T-cell responses may be context-dependent. Finally, mirroring previous findings with IL-6,114 IL-21 has been reported to render CD4+ CD25− T cells resistant to Treg-mediated suppression,115, 116 and 117 providing another mechanism through which IL-21 may exacerbate intestinal inflammation.
Taken together, the pleiotropic effects of IL-21 may contribute to chronic intestinal inflammation in two main ways. First, IL-21 may augment pathogenic leukocyte responses in the gut, especially those mediated by Th1 and Th17 cells, and potentially also by NK cells. Second, IL-21 may exacerbate inflammation through its effects on tissue cells in the gut, stimulating the production of T-cell chemoattractants and inducing the release of tissue-degrading matrix metalloproteinases. Further studies on the effects of IL-21 depletion in murine models of chronic IBD should help test these hypotheses and validate IL-21 as a possible therapeutic target.
IL-22 Plays a Key Role in Protecting the Mucosal Barrier
Another effector cytokine that is preferentially produced by Th17 cells is IL-22, which is a member of the IL-10 family of cytokines.118, 119 and 120 The IL-22R complex is not expressed by leukocytes, but is highly expressed by epithelial cells in the intestine, skin, and lung, as well as by resident tissue cells in many organs including the liver and kidney.118, 119, 120, 121, 122, 123 and 124 Thus IL-22 targets non-hematopoietic cells and has been primarily proposed to increase innate tissue defenses.122 An involvement of IL-22 in chronic intestinal inflammation is suggested by the findings that colonic and serum IL-22 levels are increased in IBD patients and in several mouse models of the disease.125, 126, 127, 128, 129 and 130 Although increased IL-22 is a feature of UC and of acute IBD models such as DSS colitis, significantly higher levels were reported in samples from CD patients and in the T-cell transfer model of colitis.125, 126, 127, 128 and 129 Recent analyses of a cohort of over 200 CD patients revealed that higher serum IL-22 levels correlated with increased disease activity and also with susceptibility-associated IL-23R polymorphisms.130 A pro-inflammatory role for IL-22 was further supported by the in vitro observations that stimulation with IL-22 led to increased expression of IL-8 and TNF-α by intestinal epithelial cells, as well as increased expression of pro-inflammatory cytokines, chemokines, and matrix metalloproteinases by colonic myofibroblasts.125, 126 However, IL-22 also stimulated β-defensin expression and increased proliferation and migration of intestinal epithelial cells, suggesting a potential role in healing and restitution of the epithelial barrier.125, 126 Very recent studies in mouse IBD models have confirmed an important role for IL-22 in protection and repair of the epithelial barrier. The UC-like pathology that spontaneously develops in TCRα−/− mice was attenuated by local delivery of IL-22, which was associated with enhanced goblet cell restitution and mucus production.129 Similarly, IL-22 expression was increased during recovery from DSS-induced acute colitis, and neutralization of IL-22 delayed recovery and goblet cell restitution.129
In addition to regulating inflammatory and repair responses, the importance of IL-22 for defense against intestinal bacterial infection was revealed in a recent study demonstrating that IL-22, but not IL-17A or IL-17F, was required for protection against infection with the intestinal bacterial pathogen C. rodentium.131 This protective IL-22 response was induced in an IL-23-dependent manner during the early phase of C. rodentium infection, but was not mediated by T cells; instead CD11c+ DC appeared to be the major source of this innate IL-22.131 Furthermore, IL-22 induced the expression of several antimicrobial proteins in the colon, including Reg family proteins, and the treatment of IL-22−/− mice with an RegIIIγ fusion protein partially restored resistance to C. rodentium infection.131 IL-22 was also recently shown to play a crucial role in protection of the lung against Klebsiella pneumoniae infection and this was associated with increased elaboration of inflammatory chemokines, as well as induction of antimicrobial peptides.124
A dual role for IL-22 in tissue protection and inflammatory pathology has also been reported in the skin. While IL-22 drives proliferation, migration, and production of antimicrobial peptides by keratinocytes,119, 122 and 132 it has also been implicated in the pathology of psoriasis lesions,120, 133 and treatment with IL-22-neutralizing antibodies ameliorated disease in a murine model.134 In contrast, studies using a mouse model of ConA-driven acute hepatitis have shown that IL-22 plays a key role in protecting against inflammatory pathology in the liver, as IL-22−/− mice exhibited greatly increased liver damage, and administration of recombinant IL-22- or IL-22-expressing Th17 cells attenuated liver pathology.135, 136 Its attenuating effects in the TCRα−/− and DSS colitis models129 illustrate a potential anti-inflammatory role for IL-22 in intestinal inflammation, and this is supported by the findings that it may stimulate IL-10 secretion by intestinal epithelial cells.123 Furthermore, some investigators have proposed that the increased serum IL-22 levels observed in CD patients may trigger an anti-inflammatory regulatory pathway through induction of lipopolysaccharide-binding protein in hepatocytes, a molecule that can limit systemic inflammatory responses to LPS.127 It remains unclear as to how IL-22 is able to mediate such divergent pro- and anti-inflammatory responses in different tissues. As IL-22R activates the STAT3 pathway, which is known to regulate both pro- and anti-inflammatory signals, additional studies of IL-22-induced STAT3 signal transduction in different cell types may provide some mechanistic insight.137
In summary, by promoting innate tissue defenses at mucosal surfaces, IL-22 plays a key role in controlling extracellular bacterial infections and in fortifying the epithelial barrier, but its role in chronic intestinal inflammation remains ambiguous. It is hard to reconcile the disease-attenuating effects of IL-22 in the DSS and TCRα−/− colitis models with its high level of expression in T-cell transfer colitis and in active CD. Further studies are required to more fully elucidate the potential role of IL-22 in chronic intestinal inflammation and to ensure that therapies targeting IL-22 in chronic disease do not result in enhanced susceptibility to mucosal infections.
Th17 and Treg Cells: Coordinating Protection and Repair Responses in the Gut?
Th17 and Treg cells are widely viewed as having a reciprocal relationship, linked through their shared use of TGF-β as a differentiation factor.54, 68, 69 and 70 However, this is an over-simplification based largely on the reported pro-inflammatory activities of Th17 cells, without full consideration of their tissue-protective functions. One possibility that has not yet been fully elucidated is that Treg and Th17 cells may play complementary roles in intestinal homeostasis. The beneficial effects of Treg activity in suppressing chronic inflammation in the gut may be partially offset by compromising protective immune responses. TGF-β plays an important role in the development and function of Treg cells and is a crucial cytokine for suppressing chronic colitis.138, 139, 140 and 141 However, in vitro studies have already indicated that Treg cells can promote the development of Th17 cells in a TGF-β-dependent manner;70, 142 therefore, the increased TGF-β associated with Treg activity may be simultaneously enhancing tissue-protective responses in the gut. It is relatively easy to imagine a scenario in which Treg cells infiltrate an inflamed gut, causing a concomitant increase in active TGF-β in an environment that is already rich in pro-inflammatory cytokines, creating favorable conditions for Th17 cell development. This effect of Treg cells could be amplified by their inhibition of Th1 responses and by their consumption of IL-2, thus reducing factors that may limit Th17 differentiation.143 It is also worth noting that the precise relationship between the Treg and the Th17 cell lineages is still unclear and there is evidence that Treg cells themselves can be induced to secrete IL-17 in vitro.142 As illustrated in Figure 1b, the synchronous activation of Treg and Th17 cells in the gut could permit the resolution of chronic inflammation by Treg cells, while maintaining Th17-mediated protective immunity, for example by increasing production of defensins and Reg proteins. In addition, as noted above, Th17-associated cytokines are also potent stimulators of mucin secretion and of intestinal epithelial cell proliferation and migration, and are therefore likely to play important roles in tissue repair and restitution of the epithelial barrier. Thus, although environments rich in IL-23 may facilitate expression of the full pathogenic potential of Th17 cells, Th17 activation in the context of a strong Treg/TGF-β response would favor tissue-protective and repair functions. In summary, the interactions between Treg and Th17 cells appear to be more complex than a simple reciprocal relationship would predict, and these distinct CD4+ T-cell subsets might work together to coordinate epithelial protection and repair in the gut.
Conclusions and Perspectives
Over the past few years, accumulating experimental and clinical data have endorsed IL-23 as a therapeutic target in IBD. However, much remains to be learned about how IL-23 drives chronic intestinal inflammation. Furthermore, the heterogeneous nature of Th17 responses, and the multiple cellular sources and functions of Th17-associated cytokines in the intestine make it difficult to precisely define the roles of individual cytokines in disease pathogenesis. Their diverse roles in coordinately regulating inflammatory as well as protective and repair responses in mucosal tissues suggest that careful consideration should be employed in weighing up the pros and cons of targeting Th17-associated cytokines in disease, a problem compounded by the heterogeneous nature of human IBD. Nevertheless, continued progress in molecular phenotyping of human IBD, in combination with further analyses of IL-23/Th17-associated cytokines in chronic IBD models, should facilitate the development of tailored therapies that target defined cytokine pathways in individual patients. Lastly, the interplay between Treg cells and the IL-23/Th17 axis in the gut is another poorly understood topic that requires further investigation. A better understanding of the IL-23/Th17 axis in intestinal homeostasis could have important implications for the rational design of future IBD therapeutics and may also open up new opportunities for improved vaccines against mucosal infections.
Disclosure
The authors declared no conflict of interest.
References
Strober, W., Fuss, I. & Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Invest. 117, 514–521 (2007).
Xavier, R.J. & Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007).
Baumgart, D.C. & Sandborn, W.J. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet 369, 1641–1657 (2007).
Scheinecker, C., Redlich, K. & Smolen, J.S. Cytokines as therapeutic targets: advances and limitations. Immunity 28, 440–444 (2008).
Oppmann, B. et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 (2000).
Kastelein, R.A., Hunter, C.A. & Cua, D.J. Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 25, 221–242 (2007).
Bouma, G. & Strober, W. The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol. 3, 521–533 (2003).
Neurath, M.F., Fuss, I., Kelsall, B.L., Stuber, E. & Strober, W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 (1995).
Simpson, S.J. et al. T cell-mediated pathology in two models of experimental colitis depends predominantly on the interleukin 12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J. Exp. Med. 187, 1225–1234 (1998).
Davidson, N.J. et al. IL-12, but not IFN-gamma, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J. Immunol. 161, 3143–3149 (1998).
Kullberg, M.C. et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66, 5157–5166 (1998).
Kullberg, M.C. et al. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 69, 4232–4241 (2001).
Monteleone, G. et al. Interleukin 12 is expressed and actively released by Crohn's disease intestinal lamina propria mononuclear cells. Gastroenterology 112, 1169–1178 (1997).
Parronchi, P. et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am. J. Pathol. 150, 823–832 (1997).
Mannon, P.J. et al. Anti-interleukin-12 antibody for active Crohn's disease. N. Engl. J. Med. 351, 2069–2079 (2004).
Fuss, I.J. et al. Both IL-12p70 and IL-23 are synthesized during active Crohn's disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 12, 9–15 (2006).
Cua, D.J. et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748 (2003).
Murphy, C.A. et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 198, 1951–1957 (2003).
Aggarwal, S., Ghilardi, N., Xie, M.H., de Sauvage, F.J. & Gurney, A.L. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278, 1910–1914 (2003).
Langrish, C.L. et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240 (2005).
Bettelli, E., Oukka, M. & Kuchroo, V.K. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 8, 345–350 (2007).
Weaver, C.T., Hatton, R.D., Mangan, P.R. & Harrington, L.E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25, 821–852 (2007).
Hue, S. et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 (2006).
Kullberg, M.C. et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203, 2485–2494 (2006).
Yen, D. et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316 (2006).
Uhlig, H. et al. Differential acitvity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25, 309–318 (2006).
Elson, C.O. et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 (2007).
Duerr, R.H. et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006).
The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Tremelling, M. et al. IL23R variation determines susceptibility but not disease phenotype in inflammatory bowel disease. Gastroenterology 132, 1657–1664 (2007).
Libioulle, C. et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 3, e58 (2007).
Rioux, J.D. et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 39, 596–604 (2007).
Glas, J. et al. rs1004819 is the main disease-associated IL23R variant in German Crohn's disease patients: combined analysis of IL23R, CARD15, and OCTN1/2 variants. PLoS ONE 2, e819 (2007).
Zhang, X.Y. et al. Identification and expression analysis of alternatively spliced isoforms of human interleukin-23 receptor gene in normal lymphoid cells and selected tumor cells. Immunogenetics 57, 934–943 (2006).
McGovern, D. & Powrie, F. The IL23 axis plays a key role in the pathogenesis of IBD. Gut 56, 1333–1336 (2007).
Eckmann, L. & Karin, M. NOD2 and Crohn's disease: loss or gain of function? Immunity 22, 661–667 (2005).
Strober, W., Murray, P.J., Kitani, A. & Watanabe, T. Signalling pathways and molecular interactions of NOD1 and NOD2. Nat. Rev. Immunol. 6, 9–20 (2006).
Burton, P.R. et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337 (2007).
Cargill, M. et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am. J. Hum. Genet. 80, 273–290 (2007).
Krueger, G.G. et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N. Engl. J. Med. 356, 580–592 (2007).
Langowski, J.L. et al. IL-23 promotes tumour incidence and growth. Nature 442, 461–465 (2006).
Goriely, S., Neurath, M.F. & Goldman, M. How microorganisms tip the balance between interleukin-12 family members. Nat. Rev. Immunol. 8, 81–86 (2008).
Re, F. & Strominger, J.L. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human dendritic cells. J. Biol. Chem. 276, 37692–37699 (2001).
Smits, H.H. et al. Commensal Gram-negative bacteria prime human dendritic cells for enhanced IL-23 and IL-27 expression and enhanced Th1 development. Eur. J. Immunol. 34, 1371–1380 (2004).
Carmody, R.J., Ruan, Q., Liou, H.C. & Chen, Y.H. Essential roles of c-Rel in TLR-induced IL-23 p19 gene expression in dendritic cells. J. Immunol. 178, 186–191 (2007).
van Beelen, A.J. et al. Stimulation of the intracellular bacterial sensor NOD2 programs dendritic cells to promote interleukin-17 production in human memory T cells. Immunity 27, 660–669 (2007).
Acosta-Rodriguez, E.V. et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 8, 639–646 (2007).
LeibundGut-Landmann, S. et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8, 630–638 (2007).
Schnurr, M. et al. Extracellular nucleotide signaling by P2 receptors inhibits IL-12 and enhances IL-23 expression in human dendritic cells: a novel role for the cAMP pathway. Blood 105, 1582–1589 (2005).
Fritz, J.H., Ferrero, R.L., Philpott, D.J. & Girardin, S.E. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 7, 1250–1257 (2006).
Kanneganti, T.D., Lamkanfi, M. & Nunez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Becker, C. et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin. Invest. 112, 693–706 (2003).
Ivanov, I.I. et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006).
Mangan, P.R. et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441, 231–234 (2006).
Denning, T.L., Wang, Y.C., Patel, S.R., Williams, I.R. & Pulendran, B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nat. Immunol. 8, 1086–1094 (2007).
Maloy, K.J. et al. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J. Exp. Med. 197, 111–119 (2003).
Izcue, A. et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28, 559–570 (2008).
Zhou, L. et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8, 967–974 (2007).
Becker, C. et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 177, 2760–2764 (2006).
Berg, D.J. et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 98, 1010–1020 (1996).
Powrie, F. et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562 (1994).
Aujla, S.J., Dubin, P.J. & Kolls, J.K. Th17 cells and mucosal host defense. Semin. Immunol. 19, 377–382 (2007).
Happel, K.I. et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 202, 761–769 (2005).
Ye, P. et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J. Exp. Med. 194, 519–527 (2001).
Wu, Q. et al. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 9, 78–86 (2007).
Khader, S.A. et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 8, 369–377 (2007).
Higgins, S.C., Jarnicki, A.G., Lavelle, E.C. & Mills, K.H. TLR4 mediates vaccine-induced protective cellular immunity to Bordetella pertussis: role of IL-17-producing T cells. J. Immunol. 177, 7980–7989 (2006).
McGeachy, M.J. & Cua, D.J. Th17 cell differentiation: the long and winding road. Immunity 28, 445–453 (2008).
Bettelli, E. et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238 (2006).
Veldhoen, M., Hocking, R.J., Atkins, C.J., Locksley, R.M. & Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006).
Harrington, L.E. et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132 (2005).
Park, H. et al. A distinct lineage of CD4 T cells regulates tissue inflam-mation by producing interleukin 17. Nat. Immunol. 6, 1133–1141 (2005).
Korn, T. et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448, 484–487 (2007).
Nurieva, R. et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448, 480–483 (2007).
Wilson, N.J. et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 8, 950–957 (2007).
McGeachy, M.J. et al. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat. Immunol. 8, 1390–1397 (2007).
Fujino, S. et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52, 65–70 (2003).
Nielsen, O.H., Kirman, I., Rudiger, N., Hendel, J. & Vainer, B. Upregulation of interleukin-12 and -17 in active inflammatory bowel disease. Scand. J. Gastroenterol. 38, 180–185 (2003).
Arisawa, T. et al. The influence of polymorphisms of interleukin-17A and interleukin-17F genes on the susceptibility to ulcerative colitis. J. Clin. Immunol. 28, 44–49 (2008).
Annunziato, F. et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 204, 1849–1861 (2007).
Kolls, J.K. & Linden, A. Interleukin-17 family members and inflammation. Immunity 21, 467–476 (2004).
Ogawa, A., Andoh, A., Araki, Y., Bamba, T. & Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 110, 55–62 (2004).
Kinugasa, T., Sakaguchi, T., Gu, X. & Reinecker, H.C. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 118, 1001–1011 (2000).
Andoh, A. et al. IL-17 selectively down-regulates TNF-alpha-induced RANTES gene expression in human colonic subepithelial myofibroblasts. J. Immunol. 169, 1683–1687 (2002).
Chen, Y. et al. Stimulation of airway mucin gene expression by interleukin (IL)-17 through IL-6 paracrine/autocrine loop. J. Biol. Chem. 278, 17036–17043 (2003).
Yang, X.O. et al. Regulation of inflammatory responses by IL-17F. J. Exp. Med. 205, 1063–1075 (2008).
Qian, Y. et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat. Immunol. 8, 247–256 (2007).
Zhang, Z., Zheng, M., Bindas, J., Schwarzenberger, P. & Kolls, J.K. Critical role of IL-17 receptor signaling in acute TNBS-induced colitis. Inflamm. Bowel Dis. 12, 382–388 (2006).
Morrissey, P.J., Charrier, K., Braddy, S., Liggitt, D. & Watson, J.D. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J. Exp. Med. 178, 237–244 (1993).
Powrie, F., Leach, M.W., Mauze, S., Caddle, L.B. & Coffman, R.L. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471 (1993).
Noguchi, D. et al. Blocking of IL-6 signaling pathway prevents CD4+ T cell-mediated colitis in a T(h)17-independent manner. Int. Immunol. 19, 1431–1440 (2007).
Monteleone, G. et al. Interleukin-21 enhances T-helper cell type I signaling and interferon-gamma production in Crohn's disease. Gastroenterology 128, 687–694 (2005).
Caruso, R. et al. IL-21 is highly produced in Helicobacter pylori-infected gastric mucosa and promotes gelatinases synthesis. J. Immunol. 178, 5957–5965 (2007).
Fina, D. et al. Interleukin-21 contributes to the mucosal T helper cell type 1 response in celiac disease. Gut 57, 887–892 (2008).
van Heel, D.A. et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat. Genet. 39, 827–829 (2007).
Leonard, W.J., Zeng, R. & Spolski, R. Interleukin 21: a cytokine/cytokine receptor system that has come of age. J. Leukoc. Biol. (2008).
Spolski, R. & Leonard, W.J. Interleukin-21: basic biology and implications for cancer and autoimmunity. Annu. Rev. Immunol. 26, 57–79 (2008).
Vollmer, T.L. et al. Differential effects of IL-21 during initiation and progression of autoimmunity against neuroantigen. J. Immunol. 174, 2696–2701 (2005).
Herber, D. et al. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J. Immunol. 178, 3822–3830 (2007).
Fina, D. et al. Regulation of gut inflammation and th17 cell response by interleukin-21. Gastroenterology 134, 1038–1048 (2008).
Parrish-Novak, J. et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 408, 57–63 (2000).
Coquet, J.M. et al. IL-21 is produced by NKT cells and modulates NKT cell activation and cytokine production. J. Immunol. 178, 2827–2834 (2007).
Harada, M. et al. IL-21-induced Bepsilon cell apoptosis mediated by natural killer T cells suppresses IgE responses. J. Exp. Med. 203, 2929–2937 (2006).
Bryant, V.L. et al. Cytokine-mediated regulation of human B cell differentiation into Ig-secreting cells: predominant role of IL-21 produced by CXCR5+ T follicular helper cells. J. Immunol. 179, 8180–8190 (2007).
Chtanova, T. et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 173, 68–78 (2004).
Caruso, R. et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology 132, 166–175 (2007).
Monteleone, G. et al. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut 55, 1774–1780 (2006).
Teramoto, K. et al. Increased lymphocyte trafficking to colonic microvessels is dependent on MAdCAM-1 and C-C chemokine mLARC/CCL20 in DSS-induced mice colitis. Clin. Exp. Immunol. 139, 421–428 (2005).
Strengell, M. et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J. Immunol. 170, 5464–5469 (2003).
Strengell, M., Sareneva, T., Foster, D., Julkunen, I. & Matikainen, S. IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J. Immunol. 169, 3600–3605 (2002).
Wurster, A.L. et al. Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J. Exp. Med. 196, 969–977 (2002).
Suto, A., Wurster, A.L., Reiner, S.L. & Grusby, M.J. IL-21 inhibits IFN-gamma production in developing Th1 cells through the repression of Eomesodermin expression. J. Immunol. 177, 3721–3727 (2006).
Pesce, J. et al. The IL-21 receptor augments Th2 effector function and alternative macrophage activation. J. Clin. Invest. 116, 2044–2055 (2006).
Pasare, C. & Medzhitov, R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science (2003).
Fantini, M.C. et al. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur. J. Immunol. 37, 3155–3163 (2007).
Peluso, I. et al. IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J. Immunol. 178, 732–739 (2007).
Clough, L.E. et al. Release from regulatory T cell-mediated suppression during the onset of tissue-specific autoimmunity is associated with elevated IL-21. J. Immunol. 180, 5393–5401 (2008).
Pestka, S. et al. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 22, 929–979 (2004).
Liang, S.C. et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 203, 2271–2279 (2006).
Zheng, Y. et al. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445, 648–651 (2007).
Dumoutier, L., Van Roost, E., Colau, D. & Renauld, J.C. Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc. Natl. Acad. Sci. USA 97, 10144–10149 (2000).
Wolk, K. et al. IL-22 increases the innate immunity of tissues. Immunity 21, 241–254 (2004).
Nagalakshmi, M.L., Rascle, A., Zurawski, S., Menon, S. & de Waal Malefyt, R. Interleukin-22 activates STAT3 and induces IL-10 by colon epithelial cells. Int. Immunopharmacol. 4, 679–691 (2004).
Aujla, S.J. et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat. Med. 14, 275–281 (2008).
Andoh, A. et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 129, 969–984 (2005).
Brand, S. et al. IL-22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G827–G838 (2006).
Wolk, K. et al. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: a potential systemic role of IL-22 in Crohn's disease. J. Immunol. 178, 5973–5981 (2007).
te Velde, A.A. et al. Comparative analysis of colonic gene expression of three experimental colitis models mimicking inflammatory bowel disease. Inflamm. Bowel Dis. 13, 325–330 (2007).
Sugimoto, K. et al. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 118, 534–544 (2008).
Schmechel, S. et al. Linking genetic susceptibility to Crohn's disease with Th17 cell function: IL-22 serum levels are increased in Crohn's disease and correlate with disease activity and IL23R genotype status. Inflamm. Bowel Dis. 14, 204–212 (2008).
Zheng, Y. et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 14, 282–289 (2008).
Boniface, K. et al. IL-22 inhibits epidermal differentiation and induces proinflammatory gene expression and migration of human keratinocytes. J. Immunol. 174, 3695–3702 (2005).
Wolk, K. et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur. J. Immunol. 36, 1309–1323 (2006).
Ma, H.L. et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J. Clin. Invest. 118, 597–607 (2008).
Radaeva, S., Sun, R., Pan, H.N., Hong, F. & Gao, B. Interleukin 22 (IL-22) plays a protective role in T cell-mediated murine hepatitis: IL-22 is a survival factor for hepatocytes via STAT3 activation. Hepatology 39, 1332–1342 (2004).
Zenewicz, L.A. et al. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity 27, 647–659 (2007).
O’Shea, J.J. & Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 28, 477–487 (2008).
Chen, W. et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 198, 1875–1886 (2003).
Marie, J.C., Letterio, J.J., Gavin, M. & Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 201, 1061–1067 (2005).
Fahlen, L. et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J. Exp. Med. 201, 737–746 (2005).
Li, M.O., Wan, Y.Y., Sanjabi, S., Robertson, A.K. & Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 24, 99–146 (2006).
Xu, L., Kitani, A., Fuss, I. & Strober, W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J. Immunol. 178, 6725–6729 (2007).
Laurence, A. et al. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26, 371–381 (2007).
Yao, Z. et al. Human IL-17: a novel cytokine derived from T cells. J. Immunol. 155, 5483–5486 (1995).
Fossiez, F. et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 183, 2593–2603 (1996).
Infante-Duarte, C., Horton, H.F., Byrne, M.C. & Kamradt, T. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 165, 6107–6115 (2000).
Stark, M.A. et al. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 22, 285–294 (2005).
Happel, K.I. et al. Cutting edge: roles of Toll-like receptor 4 and IL-23 in IL-17 expression in response to Klebsiella pneumoniae infection. J. Immunol. 170, 4432–4436 (2003).
Tajima, M. et al. IL-6-dependent spontaneous proliferation is required for the induction of colitogenic IL-17-producing CD8+ T cells. J. Exp. Med. 205, 1019–1027 (2008).
Lockhart, E., Green, A.M. & Flynn, J.L. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177, 4662–4669 (2006).
Yang, X.O. et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28, 29–39 (2008).
Pichavant, M. et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J. Exp. Med. 205, 385–393 (2008).
Rachitskaya, A.V. et al. Cutting edge: NKT cells constitutively express IL-23 receptor and RORgammat and rapidly produce IL-17 upon receptor ligation in an IL-6-independent fashion. J. Immunol. 180, 5167–5171 (2008).
Lee, K.A. et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell Immunol. (2008).
Ferretti, S., Bonneau, O., Dubois, G.R., Jones, C.E. & Trifilieff, A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J. Immunol. 170, 2106–2112 (2003).
Veldhoen, M. et al. The aryl hydrocarbon receptor links T(H)17-cell-mediated autoimmunity to environmental toxins. Nature (2008).
Suzuki, S. et al. Expression of interleukin-17F in a mouse model of allergic asthma. Int. Arch. Allergy Immunol. 143 (Suppl 1), 89–94 (2007).
Liang, S.C. et al. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J. Immunol. 179, 7791–7799 (2007).
Wright, J.F. et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J. Biol. Chem. 282, 13447–13455 (2007).
Chang, S.H. & Dong, C. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res. 17, 435–440 (2007).
Chung, Y. et al. Expression and regulation of IL-22 in the IL-17-producing CD4+ T lymphocytes. Cell Res. 16, 902–907 (2006).
Acknowledgements
KM is supported by the Wellcome Trust. MK is supported by grants from the Wellcome Trust and the Medical Research Council.
Author information
Authors and Affiliations
Corresponding authors
PowerPoint slides
Rights and permissions
About this article
Cite this article
Maloy, K., Kullberg, M. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol 1, 339–349 (2008). https://doi.org/10.1038/mi.2008.28
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2008.28
This article is cited by
-
Immune Inhibitory Properties and Therapeutic Prospects of Transforming Growth Factor-Beta and Interleukin 10 in Autoimmune Hepatitis
Digestive Diseases and Sciences (2022)
-
Macrophage polarization in intestinal inflammation and gut homeostasis
Inflammation Research (2020)
-
Alcohol-induced IL-17A production in Paneth cells amplifies endoplasmic reticulum stress, apoptosis, and inflammasome-IL-18 activation in the proximal small intestine in mice
Mucosal Immunology (2019)
-
IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting
Nature Reviews Gastroenterology & Hepatology (2019)
-
Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis
Mucosal Immunology (2019)
