
Abstract
ARCTIC was a multicenter, randomized-controlled, open, phase IIB non-inferiority trial in previously untreated chronic lymphocytic leukemia (CLL). Conventional frontline therapy in fit patients is fludarabine, cyclophosphamide and rituximab (FCR). The trial hypothesized that including mitoxantrone with low-dose rituximab (FCM-miniR) would be non-inferior to FCR. A total of 200 patients were recruited to assess the primary end point of complete remission (CR) rates according to IWCLL criteria. Secondary end points were progression-free survival (PFS), overall survival (OS), overall response rate, minimal residual disease (MRD) negativity, safety and cost-effectiveness. The trial closed following a pre-planned interim analysis. At final analysis, CR rates were 76 FCR vs 55% FCM-miniR (adjusted odds ratio: 0.37; 95% confidence interval: 0.19–0.73). MRD-negativity rates were 54 FCR vs 44% FCM-miniR. More participants experienced serious adverse reactions with FCM-miniR (49%) compared to FCR (41%). There are no significant differences between the treatment groups for PFS and OS. FCM-miniR is not expected to be cost-effective over a lifetime horizon. In summary, FCM-miniR is less well tolerated than FCR with an inferior response and MRD-negativity rate and increased toxicity, and will not be taken forward into a confirmatory trial. The trial demonstrated that oral FCR yields high response rates compared to historical series with intravenous chemotherapy.
Similar content being viewed by others


Introduction
Chronic lymphocytic leukemia (CLL) is the commonest leukemia above the age of 50 years with a median age of diagnosis of 70 years.
CLL is an incurable disease, and most patients will eventually become resistant to the treatment. For physically fit patients, the addition of rituximab (MabThera) to fludarabine and cyclophosphamide (FCR) has become the standard of care based on evidence from large randomized-controlled trials1, 2 and large single-center experience.3 However, the dose of rituximab has not been established systematically in CLL. Rituximab monotherapy at a dose of 375 mg/m2 induced an overall response rate (ORR) of 13% in previously-treated CLL/small lymphocytic lymphoma.4 Thrice weekly rituximab (375 mg/m2) and higher weekly doses of rituximab (0.5–2.5 g/m2) in previously untreated patients induced a modest ORR of 43% and 40%, respectively.5, 6, 7 The poor response was thought to be due to low CD20 expression on CLL cells and rituximab binding to CD20 positive cellular debris. The loss of CD20 antigen from CLL cells when exposed to rituximab (termed ‘antigen shaving’) is well described. Most of the cells were cleared after 30 mg of rituximab followed by recrudescence of CLL cells that have lost >90% of CD20 expression. Low-dose rituximab thrice weekly at 20–60 mg/m2 may promote enhanced clearance of CLL cells by preserving CD20 expression.8, 9 Subcutaneous rituximab thrice weekly at a dose of 20 mg resulted in reduction of CD20 expression on CLL cells, but sufficient expression was maintained during the course of 6–12 weeks in another study.10 Thrice weekly rituximab at 20 mg/m2 in combination with Alemtuzumab and Pentostatin showed that this dose is able to opsonize and clear the majority of circulating cells, but the loss of CD20 is less pronounced.11 Hence, rituximab at doses of 20 mg/m2 can be effective in CLL.
Mitoxantrone in combination with fludarabine and cyclophosphamide (FCM) has been reported to give an ORR of 78% in patients with relapsed or resistant CLL.12 The combination of chemotherapy with rituximab (FCM-R) has been used in previously untreated CLL patients in phase II trials with good response rates.13, 14
The aim of the attenuated dose rituximab with chemotherapy in CLL (ARCTIC) trial was to test the hypothesis that low-dose of rituximab (100 mg per cycle) in combination with FCM (FCM-miniR) would be as effective as standard of care (FCR). It is hypothesized that FCM-miniR may result in effective tumor clearance and preservation of CD20 expression on CLL cells.
The cost-effectiveness of delivering FCM-miniR as an alternative to the standard therapy FCR is also critical. Six cycles of rituximab at a dose of 500 mg/m2 are time-consuming to give and expensive compared to low doses (100 mg per cycle). The non-inferiority design helps to establish whether lowering the dose of rituximab and hence reducing the cost of treatment impacts on the efficacy in terms of CR rates, as well as the longer-term progression-free survival (PFS) and overall survival (OS) outcomes.
ARCTIC ran concurrently with the ADMIRE trial,15 which aimed to assess whether the addition of mitoxantrone to FCR (FCM-R) increased the depth of response compared to standard treatment with FCR. Both trials recruited from the same population of patients but from different centers within the UK.
Patients and methods
Trial design and patients
ARCTIC was a multicenter, randomized, controlled, open-label, phase IIB non-inferiority trial including patients with previously untreated CLL who required treatment by IWCLL criteria.16 Patients were randomized via a central computer-generated minimization programme incorporating a random element 1:1 to FCR or FCM-miniR. Randomization was stratified to ensure balance for center, Binet stage (progressive A or B, C), age group (⩽65, 65) and sex. The intention was to recruit 206 patients from hospitals around the United Kingdom. All participants provided written informed consent. Detailed eligibility criteria is provided in the Supplementary Information (SI).
The primary objective was to assess whether FCM-miniR was non-inferior to FCR in terms of CR rates, including CR with incomplete marrow recovery (CRi), in patients with previously untreated CLL. The results would be used to determine whether FCM-miniR should be taken forward into a larger definitive phase III trial.
An independent Data Monitoring Committee (DMC) was established to review the safety and ethics of the trial. There was a pre-planned interim assessment of efficacy on half the required number of participants. The DMC reported to an established trial steering committee (TSC) that provided general oversight for the trial.
The trial protocol was approved by the Leeds East Research Ethics Committee and the Medicine and Healthcare products Regulatory Agency (MHRA). The trial was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. The trial was registered as an International Standard Randomized Controlled Trial (ISRCTN16544962) and on the European Clinical Trials Database (EudraCT: 2009-010998-20).
Treatment and assessments
Treatment with FCR or FCM-miniR was repeated every 28 days for a total of six cycles. Fludarabine and cyclophosphamide were administered orally at doses which are pharmacologically equivalent to the doses used when FCR is given intravenously for CLL.17 This is in contrast to similar studies where intravenous doses of fludarabine and cyclophosphamide are used.1, 2, 3, 12 Full details of both treatment schedules are provided in the SI.
Participants were assessed for response to treatment at 3 months post treatment, 12, 18 and 24 months post randomization, or until disease progression requiring treatment. Long-term annual follow-up for survival is performed until death.
End points
The primary end point was the CR rate (including CRi) at 3 months post treatment. A central blinded assessment of response was conducted according to the IWCLL criteria16 by two independent CLL hematologists. An independent arbiter reviewed discordant reports.
Secondary end points at 3 months post treatment included MRD negativity, assessed in the bone marrow by multiparameter flow cytometry with a level of detection below 1 CLL cell in 10 000 leukocytes;18, 19 ORR defined as at least a partial remission (PR); safety and toxicity as graded by CTCAE V3.0.20
Longer-term secondary end points included PFS, OS, time to MRD relapse in participants who became MRD negative and cost-effectiveness.
Sample size
Previous studies showed FCR CR rates of at least 50%.2, 21 With 80% power to show non-inferiority, where this is defined as FCM-miniR being not >10% worse in terms of CR rates than FCR, an assumed 10% difference in favor of FCM-miniR, a one-sided significance level of 2.5%22 and 80% power, 98 patients were required per group. A total of 206 patients were planned, allowing for 5% dropout.
A formal interim analysis to allow large treatment group differences to be reported early was planned on the short-term efficacy data on half the required participants (n=103). A stringent significance level was required for the interim analysis (0.005, two-sided) using the O’Brien–Fleming23 alpha-spending function.
Statistical methods
All analyses were conducted on the intention-to-treat population, in which participants were included according to their randomized treatment. A per-protocol analysis was planned for the primary end point, including participants who received at least one cycle of treatment as protocoled and were not major eligibility violators. Safety analyses included participants according to the treatment they actually received.
Methods for handling missing end point data were prespecified and approved by the Chief Investigator. Participants with a missing assessment who died from CLL or treatment-related toxicity prior to their primary end point assessment, or discontinued treatment early due to non-response or toxicity were treated as non-responders/MRD positive. In the formal statistical analysis of the primary end point, for participants with at least a PR but missing trephine data to confirm a CR, imputation methods treated MRD-negative participants as having a CR and MRD positive as not, although summaries also report the un-imputed data. Participants without an available end point assessment, were not included in the formal statistical analysis of the primary end point. This was appropriate as it can be assumed that data are missing completely at random, since assessments were most likely unavailable due to samples being un-assessable or missed in error, rather than participant refusal due to level of response or treatment allocation. Sensitivity analyses assessed the robustness of the assumptions regarding missing primary end point data.
Multivariable binary logistic regression models compared CR rates, proportions with undetectable MRD (MRD negative) and ORR between the treatment groups, adjusting for the minimization factors, excluding center. Parameter estimates, s.e. and odds ratios (ORs) for the treatment effect are reported. The differences in proportions are reported with 95% confidence intervals (CIs). The lower limit of the CI for the CR rates was compared with the non-inferiority margin of 10%, expressed as an OR.
Kaplan–Meier curves are presented for the PFS and OS end points. Multivariable Cox regression analysis formally compared time to MRD relapse, PFS and OS between the treatment groups, hazard ratios (HRs), 95% CIs and corresponding P-values for the treatment effect are reported. Participants without evidence of an event at the time of analysis were censored at the last date they were known to be alive and event-free.
Safety analyses summarized the number of safety events occurring after randomization including treatment-related mortalities and incidence of secondary cancers.
Prespecified exploratory subgroup analyses assessed the heterogeneity of the treatment effect among subgroups of interest for the primary end point, PFS and OS. Formal statistical testing between subgroups was not appropriate due to multiple testing errors and the reduced numbers in each subgroup.
An economic evaluation was conducted from a National Health Service (NHS) and Personal Social Services (PSS) perspective, with health benefit measured in quality-adjusted life years (QALYs), using patient-reported EQ-5D-3L questionnaires.24 A within-trial analysis compared the outcomes and costs over 24 months using individual patient data from the trial, and a modified Markov model was used to estimate lifetime cost-effectiveness. The model included three health states: disease free, recurrence and death. Results are reported in 2013 GBP (£), and for information costs are presented in US dollars ($) using an exchange rate of 1:1.43.
Code availability
All statistical analyses were carried out using SAS software 9.4 (SAS Institute, Cary, NC, USA). Statistical analysis programs were validated internally but are not available publicly.
Results
Recruitment and early closure
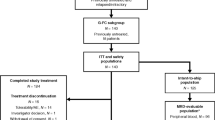
The CONSORT diagram (Figure 1) shows the flow of participants throughout the trial. A total of 548 patients were screened for eligibility, and 200 randomized. Of the 348 patients not randomized, the majority were clinically ineligible (n=228, 65.5%). Common reasons included: asymptomatic CLL, poor performance status, prior therapy for CLL, second malignancy or not having B-cell chronic lymphocytic leukemia.

CONSORT diagram.
At the pre-planned interim analysis on the first 103 participants, 72 (69.9%) received six cycles of treatment (FCR: 38/51 (74.5%), FCM-miniR: 34/52 (65.4%)) and 61/103 (59.2%) achieved a CR (FCR: 34/51 (66.7%), FCM-miniR: 27/52 (51.9%)). Removing those without an assessable response, 61/85 (71.8%) achieved a CR (FCR: 34/41 (82.9%), FCM-miniR: 27/44 (61.4%)), with a difference in response rates (FCM-miniR–FCR) of −21.6% (99.5% CI: −48.0, 4.8%), adjusted P=0.037. Although not significant at the pre-planned interim level (α=0.005), the results approached significance in favor of FCR and were deemed clinically significant by the DMC. There was also evidence of additional toxicity in the FCM-miniR group with 65.4% (34/52) experiencing a serious adverse event compared to 51.0% (26/51) with FCR. The DMC and TSC recommended closure to recruitment with immediate effect. Any participants still receiving FCM-miniR were recommended to crossover to FCR (labeled FCM-miniR/FCR). A total of 21/23 participants switched treatment. By the time of the interim analysis, and the 103 participants had been followed up for their primary end point and the analysis conducted, 200 participants had been recruited. Participants were randomized between December 2009 and September 2012 (FCR:100, FCM-miniR:100) from 34 UK institutions with local ethical and management approval. At the time of reporting, it has been approximately 6 years since the trial opened to recruitment, with a median follow-up of just over 4 years.
Patient characteristics
Baseline characteristics are displayed in Table 1. The median age was 63 years (range 36–80) with 75 participants (37.5%) aged >65 years. There was a male predominance (135 (67.5%)), and 34 participants (17.0%) were Binet stage progressive A, 95 (47.5%) stage B and 71 (35.5%) stage C. A majority of participants (116 (58.0%)) were World Health Organization performance status 0, with 77 (38.5%) performance status 1 and 7 (3.5%) performance status 2. Overall, 103 participants (51.5%) had B-symptoms, a higher proportion with FCM-miniR (FCR: 46 (46.0%), FCM-miniR: 57 (57.0%)), while 115 (57.5%) had a β2-microglobulin concentration of ⩾4 mg/l and 31 (15.5%) creatinine clearance levels of 30–60 mls/min. Of the evaluable participants, 7/183 (3.8%) had a 17p deletion (FCR: 4 (4.3%), FCM-miniR: 3 (3.3%)) and 30/188 (16.0%) an 11q deletion (FCR: 10 (10.8%), FCM-miniR: 20 (21.1%)). A total of 104/165 participants (63.0%) were considered to be ‘poorer risk’ (FCR: 52 (63.4%), FCM-miniR: 52 (62.7%)), in terms of VH mutational status, that is, VH unmutated or involving the VH3-21 gene. Sixteen participants (9.7%) presented with the VH3-21 gene (FCR: 7; FCM-miniR: 9).
Treatment
Of the 200 participants, 141 (70.5%) received six cycles of treatment (FCR: 70 (70.0%), FCM-miniR: 51 (64.5%), FCM-miniR/FCR: 20 (95.2%)) and 31 (15.5%) received ⩽3 cycles of treatment (FCR: 15 (15.0%), FCM-miniR: 16 (20.3%), FCM-miniR/FCR: 0 (0.0%)) (Table 2). Two FCR participants did not receive any trial treatment, one had received prior therapy for CLL, and one had a 17p deletion and was withdrawn from the trial (Figure 1). Overall, 59 participants (29.5%) discontinued treatment prematurely (FCR: 30 (30.0%), FCM-miniR: 28 (35.4%), FCM-miniR/FCR: 1(4.8%)). Reasons included: toxicity (n=44); progressive disease (n=3); stable disease with no or minimal response (n=3); ineligibility (n=1), patient decision (n=3); clinician decision (n=4); and other (n=1). A total of 94 participants (47.0%) received granulocyte-colony stimulating factor (G-CSF) during treatment as recommended in the protocol as secondary prophylaxis, with a higher proportion in the FCM-miniR group (FCR: 42 (42.0%), FCM-miniR: 40 (50.6%)) (Table 2). Thirteen participants unable to tolerate oral chemotherapy received equivalent intravenous doses (FCR: 7 (7.0%), FCM-miniR: 5 (6.3%), FCM-miniR/FCR: 1 (4.8%)).
Efficacy
Of the 200 participants, 124 (62.0%) achieved a CR (FCR: 68 (68.0%), FCM-miniR: 39 (49.4%), FCM-miniR/FCR: 17 (81.0%)). In the formal analysis of the primary end point after imputation using MRD outcome, 111/167 (66.5%) achieved a CR, (FCR: 70/92 (76.1%), FCM-miniR: 41/75 (54.7%)). The difference in response rates (FCM-miniR–FCR) was −21.4% in favor of FCR (95% CI: −35.8, −7.0%). In the multivariable logistic regression analysis, the OR for achieving a CR with FCM-miniR compared to FCR was 0.37 (95% CI: 0.19, 0.73; Table 3). A 10% non-inferiority reduction from the FCR CR rate gives an OR limit of 0.61. Since the lower limit, and in fact the mean of the 95% CI for the treatment effect is <0.61, and the upper limit is below 1, there is evidence that FCM-miniR is significantly inferior to FCR. The per-protocol analysis (n=166) concurred with the outcome of the intention-to-treat analysis, OR=0.38 (95% CI: 0.19, 0.75). The sensitivity analyses (including comparison of CR rates prior to imputation) did not alter the findings.
There were no large differences in proportions achieving a CR by sex (males: 76/117 (65.0%), females: 35/50 (70.0%)), age group (⩽65: 75/106 (70.8%), >65: 36/61 (59.0%)), or Binet stage (A progressive/B: 76/111 (68.5%), C: 35/56 (62.5%)) or creatinine clearance levels (mls/min) (30–60: 15/25 (60.0%), >60: 96/142 (67.6%)). A significantly higher proportion of participants who received more than three cycles of treatment achieved a CR (⩽3 cycles: 7/25 (28.0%), >3 cycles: 104/142 (73.2%)), with difference (−45.2% (95% CI: −64.3, −26.2%)). The achievement of a CR was 6.2% lower for those participants who received G-CSF during treatment cycles two to six than for those who did not (G-CSF received: 50/77 (64.9%), no G-CSF: 59/83 (71.1%)).
All assessable participants with a 17p deletion failed to achieve a CR (n=6). Lower proportions of participants with an 11q deletion and ‘poorer risk’ VH mutational status achieved a CR (11q deletion: 14/24 (58.3%), no 11q deletion: 90/133 (67.7%)), (VH unmutated or VH3-21: 54/87 (62.1%), VH mutated: 36/52 (69.2%)).
Of the 200 participants, 184 (92.0%) achieved at least a PR (FCR: 94 (94.0%), FCM-miniR: 69 (87.3%), FCM-miniR/FCR: 21 (100%)). Of the assessable participants, the ORR was 92.6% (163/176) with a higher proportion in the FCR group than FCM-miniR (FCR: 94/98 (95.9%), FCM-miniR: 69/78 (88.5%)), with a difference (FCM-miniR–FCR) of −7.5% (95% CI: −15.6%, 0.6%)). A binary logistic regression analysis formally comparing the ORR between the treatment groups was unable to be performed due to the small number of participants in the non-responders group.
Of the 200 participants, 85 (42.5%) achieved MRD negativity assessed in the bone marrow at 3 months post treatment (FCR: 45 (45.0%), FCM-miniR: 29 (36.7%), FCM-miniR/FCR: 11 (52.4%)). In the formal analysis of MRD (excluding FCM-miniR/FCR participants and those with a missing MRD assessment), 74/149 (49.7%) achieved MRD negativity (FCR: 45 (54.2%), FCM-miniR: 29 (43.9%)). The difference in response rates (FCM-miniR–FCR) was −10.3% (95% CI: −26.3, 5.8%), P-value=0.213. In the multivariable logistic regression analysis, the adjusted OR for achieving MRD negativity with FCM-miniR compared to FCR was 0.65 (95% CI: (0.33, 1.26), P=0.198) concluding that there is no evidence of a significant difference between the treatment groups (Table 3).
At the time of analysis (3 years post randomization of the final participant), 33 (16.5%) participants have died (FCR: 14 (14.0%), FCM-miniR: 18 (22.8%), FCM-miniR/FCR: 1 (4.8%)), and 73 (36.5%) have either progressed or died (FCR: 34 (34.0%), FCM-miniR: 35 (44.3%), FCM-miniR/FCR: 4 (19.0%)). Table 4 presents the primary cause of death by treatment group. Of the 33 participant deaths, 18 (54.5%) were due to CLL, that is, infection due to CLL, overwhelming tumor load or high-grade transformation of CLL (FCR: 5 (35.7%), FCM-miniR: 13 (72.2%)). Seven (21.2%) were treatment-related including treatment-related MDS/AML and infection due to treatment (FCR: 4 (28.6%), FCM-miniR: 2 (11.1%), FCM-miniR/FCR: 1 (100%)). Figure 2 presents the PFS and OS Kaplan–Meier curves by treatment group (excluding FCM-miniR/FCR participants). At 36 months post randomization, the PFS rate is FCR: 75.3 vs FCM-miniR: 71.3%; with OS rate FCR: 89.1%, FCM-miniR: 84.3%. The HRs were not significant in the adjusted Cox regression model (PFS: HR=1.29, 95% CI: (0.80, 2.07), P=0.298; OS: HR=1.62, 95% CI: (0.80, 3.28), P=0.178).

Kaplan–Meier curves for progression-free and overall survival. (a) Progression-free survival by treatment group. (b) Overall survival by treatment group.
Of the 85 participants who were MRD negative in the bone marrow at 3 months post treatment (Table 3), 9 (10.6%) were reported to have relapsed at the MRD level in the peripheral blood or progressed (FCR: 5/45 (11.1%), FCM-miniR: 4/29 (13.8%)) at the end of the planned 2-year follow-up. The curves are not presented due to the small number of events.
For the planned subgroup analyses, Kaplan–Meier curves demonstrated an improved PFS in participants who achieved a CR or MRD negativity at 3 months post treatment (Figure 3). There was a trend toward participants with a VH mutated gene (and not VH3-21), that is, ‘standard risk’ patients showing an improved PFS over those with a ‘poor risk’ (Figure 3). Sensitivity analyses for both CR status prior to imputation and subgroup analyses for OS show similar trends.

Kaplan–Meier curves for progression-free survival subgroup analyses. (a) PFS by CR status at 3 months post treatment. (b) PFS by MRD status at 3 months post treatment (assessed in the bone marrow). (c) PFS by VH mutational risk status.
Economic evaluation
Over the planned 24-month trial period, FCM-miniR produced a mean cost saving of £6 619 ($9649) (s.d. £1061 ($1518)) and QALY loss of −0.059 (s.d. 0.06) compared to FCR. Assuming that one QALY is valued at £20 000, as per UK standard, FCM-miniR is cost-effective over the trial period, producing a positive incremental net health benefit (+0.27 QALYs; s.d. 0.08) due to the short-term cost savings associated with FCM-miniR treatment. However, FCM-miniR is not expected to be cost-effective over a lifetime horizon, with an expected lifetime cost saving of £7723 ($11 048) (s.d. £3281 ($4694)), and QALY loss of −0.73 (s.d. 0.42), resulting in an incremental net health loss (QALY: −0.34; s.d. 0.40; Table 5).
Safety and toxicity
The safety population included 198 participants (Figure 1). A total of 183 serious adverse events (SAEs) were reported from 104 (52.5%) participants, from a lower proportion receiving FCR (49.0%) compared to FCM-miniR (58.2%). A total of 145 serious adverse reactions were reported from 89 (44.9%) participants (FCR: 62 events from 41 (41.0%); FCM-miniR: 67 events from 39 (49.4%); FCM-miniR/FCR: 16 events from 9 (47.4%)). The most commonly reported serious adverse reactions (SARs), 62.1% of events (n=90), were infections and infestations. Ninety-six (48.5%) participants required hospitalization for an SAE with similar proportions in each treatment group (Table 6).
One suspected unexpected serious adverse reaction (SUSAR) was reported from a participant receiving FCR. A squamous cell carcinoma, two lesions on the lower back and central chest was diagnosed approximately 4 months after the participant received six cycles of treatment.
Non-serious adverse events (AEs) were reported from 192 (97.0%) participants with similar proportions in each treatment group. Of the 2163 AEs reported, 388 (17.9%) were graded as CTCAE grade 3 or above (FCR: 168 (15.0%); FCM-miniR: 193 (22.4%); FCM-miniR/FCR: 27 (14.8%)) (Table 6). There were 339 reports of neutropenia (FCR: 157 (14.1%), FCM-miniR: 152 (17.6%), FCM-miniR/FCR: 30 (16.4%)) and 190 reports of thrombocytopenia (FCR: 91 (8.1%), FCM-miniR: 89 (10.3%), FCM-miniR/FCR: 10 (5.5%)) during the course of the trial.
There were no treatment-related mortalities reported within 3 months of the end of protocol treatment.
Within 4 years following treatment, 26 participants (13.1%) had been diagnosed with a second cancer (FCR: 13 (13.0%); FCM-miniR: 12 (15.2%); FCM-miniR/FCR: 1 (5.3%)). The most common were non-melanoma skin cancers in 5.1% (n=10) of participants, followed by hematological cancers (AML/MDS) in 3.0% (n=6) (Table 6). There have been five reports of myelodysplastic syndrome (MDS) (FCR:2, FCM-miniR:3).
Discussion
Participants randomized to FCM-miniR had a significantly lower CR rate than those randomized to FCR (54.7% vs 76.1%), indicating that FCR is the more effective treatment. This seems, at least in part, due to the higher toxicity associated with the addition of mitoxantrone to FCR with 41.1% of participants receiving FCR reporting a SAR compared with 49.4% receiving FCM-miniR. The increase in toxicity observed with FCM-miniR compared to FCR was similar to the increase seen with FCM-R compared to FCR in the companion ADMIRE study.15 This would suggest that additional toxicity is likely to be related to addition of mitoxantrone. Key secondary end points were consistent in demonstrating that FCR has greater efficacy, with a higher proportion of participants achieving MRD negativity (FCR: 54.2%, FCM-miniR: 43.9%). Trial follow-up is at a median of 4 years, and there are a high number of censored observations, but to date the PFS and OS are favorable compared to previous studies. There are no significant differences between the treatment groups for PFS and OS.
The cost-effectiveness analysis indicates that while FCM-miniR is expected to be cost-effective in the short term, it is unlikely to be cost-effective when taking into account long-term costs and health benefits, although there is significant uncertainty around the long-term results.
The design of this trial and its companion trial, ADMIRE comparing FCR with FCM-R,15 were based on several non-randomized phase II trials suggesting that the addition of mitoxantrone to FCR improved outcomes in CLL. The lower dose of rituximab was based on pre-clinical and biological responses seen in small studies examining the impact of lower doses of rituximab as a single agent in CLL. Both trials failed to demonstrate the expected improvement in outcome for the proposed interventions. The use of randomized phase II trials allows a more critical assessment of the value of any proposed changes to treatment giving a more robust assessment prior to launching prolonged and expensive phase III trials. Given the rapidly changing therapy in diseases such as CLL, the use of randomized phase II trials either as stand-alone trials or as part of seamless phase II/III designs is an efficient way to prioritize appropriate phase III trial design and is highly recommended compared to large non-comparative phase II trials that are commonly performed.
In addition the outcomes for both the ARCTIC and ADMIRE15 trials are consistent with each other and demonstrate that the delivery of fludarabine and cyclophosphamide orally in FCR is at least as effective as, and possibly more effective than, FCR when F and C are given intravenously; although we acknowledge the lack of head-to-head comparisons in this area. Oral FCR is also much more convenient for patients and results in less use of valuable medical resources as patients only require a single day case visit per cycle of treatment rather than three that is required if FCR is given intravenously.
In summary, we demonstrate that FCM-miniR is not non-inferior to FCR in terms of the primary end point of CR at 3 months post treatment. In addition, FCM-miniR shows evidence of reduced efficacy in terms of MRD and survival, had increased toxicity, and is not cost-effective longer term. In view of this, FCM-miniR will not be taken forward into a larger definitive phase III trial. The trial demonstrated that oral FCR yields extremely high response and MRD-negativity rates compared to historical series in which the chemotherapy was given intravenously, and remains the gold-standard therapy for CLL in participants considered fit for fludarabine-based therapy. The addition of subcutaneous rituximab to oral FC should be explored in future trials to make this therapy more adaptable to patients’ need without compromising efficacy. We also demonstrate the value of randomized phase II trials to improve the quality of future phase III trials.
References
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood 2016; 127: 208–215.
Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010; 376: 1164–1174.
Tam CS, O'Brien S, Wierda W, Kantarjian H, Wen S, Do K-A et al. Long-term results of the fludarabine, cyclophosphamide, and rituximab regimen as initial therapy of chronic lymphocytic leukemia. Blood 2008; 112: 975–980.
McLaughlin P, Grillo-López AJ, Link BK, Levy R, Czuczman MS, Williams ME et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16: 2825–2833.
Keating M, O'Brien S . High-dose rituximab therapy in chronic lymphocytic leukemia. Semin Oncol 2000; 27 (6 Suppl 12): 86–90.
O’Brien SM, Kantarjian H, Thomas DA, Giles FJ, Freireich EJ, Cortes J et al. Rituximab dose-escalation trial in chronic lymphocytic leukemia. J Clin Oncol 2001; 19: 2165–2170.
Byrd JC, Murphy T, Howard RS, Lucas MS, Goodrich A, Park K et al. Rituximab using a thrice weekly dosing schedule in B-cell chronic lymphocytic leukemia and small lymphocytic lymphoma demonstrates clinical activity and acceptable toxicity. J Clin Oncol 2001; 19: 2153–2164.
Williams ME, Densmore JJ, Pawluczkowycz AW, Beum PV, Kennedy AD, Lindorfer MA et al. Thrice-weekly low-dose rituximab decreases CD20 loss via shaving and promotes enhanced targeting in chronic lymphocytic leukemia. J Immunol 2006; 177: 7435–7443.
Almasri NM, Duque RE, Iturraspe J, Everett E, Braylan RC . Reduced expression of CD20 antigen as a characteristic marker for chronic lymphocytic leukemia. Am J Hematol 1992; 40: 259–263.
Aue G, Lindorfer MA, Beum PV, Pawluczkowycz AW, Vire B, Hughes T et al. Fractionated subcutaneous rituximab is well-tolerated and preserves CD20 expression on tumor cells in patients with chronic lymphocytic leukemia. Haematologica 2010; 95: 329–332.
Zent CS, Taylor RP, Lindorfer MA, Beum PV, LaPlant B, Wu W et al. Chemoimmunotherapy for relapsed/refractory and progressive 17p13-deleted chronic lymphocytic leukemia (CLL) combining pentostatin, alemtuzumab, and low-dose rituximab is effective and tolerable and limits loss of CD20 expression by circulating CLL cells. Am J Hematol 2014; 89: 757–765.
Bosch F, Ferrer A, Villamor N, González M, Briones J, González-Barca E et al. Fludarabine, cyclophosphamide, and mitoxantrone as initial therapy of chronic lymphocytic leukemia: high response rate and disease eradication. Clin Cancer Res 2008; 14: 155–161.
Bosch F, Abrisqueta P, Villamor N, Terol MJ, González-Barca E, Ferra C et al. Rituximab, fludarabine, cyclophosphamide, and mitoxantrone: a new, highly active chemoimmunotherapy regimen for chronic lymphocytic leukemia. J Clin Oncol 2009; 27: 4578–4584.
Hillmen P, Cohen DR, Cocks K, Pettitt A, Sayala HA, Rawstron AC et al. A randomized phase II trial of fludarabine, cyclophosphamide and mitoxantrone (FCM) with or without rituximab in previously treated chronic lymphocytic leukaemia. Br J Haematol 2011; 152: 570–578.
Munir T, Howard DR, McParland L, Pocock C, Rawstron AC, Hockaday A et al. Results of the randomized phase IIB ADMIRE trial of FCR with or without mitoxantrone in previously untreated CLL. Leukemia 2017; (in press).
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on chronic lymphocytic leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008; 111: 5446–5456.
Dearden CE, Richards S, Else M, Catovsky D, Hillmen P . A comparison of the efficacy and safety of oral and intravenous fludarabine in chronic lymphocytic leukemia in the LRF CLL4 trial. Cancer 2011; 117: 2452–2460.
Rawstron AC, Fazi C, Agathangelidis A, Villamor N, Letestu R, Nomdedeu J et al. A complementary role of multiparameter flow cytometry and high-throughput sequencing for minimal residual disease detection in chronic lymphocytic leukemia: an European Research Initiative on CLL study. Leukemia 2016; 30: 929–936.
Rawstron AC, Bottcher S, Letestu R, Villamor N, Fazi C, Kartsios H et al. Improving efficiency and sensitivity: European Research Initiative in CLL (ERIC) update on the international harmonised approach for flow cytometric residual disease monitoring in CLL. Leukemia 2013; 27: 142–149.
Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events, 2003. Available at: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf.
Hallek M, Fingerle-Rowson G, Fink A-M, Busch R, Mayer J, Hensel M et al. Immunochemotherapy with fludarabine (F), cyclophosphamide (C), and rituximab (R) (FCR) versus fludarabine and cyclophosphamide (FC) improves response rates and progression-free survival (PFS) of previously untreated patients (pts) with advanced chronic lymphocytic leukemia (CLL). ASH Annual Meeting Abstracts 2008; 112 (11): 325.
Kay R. Equivalence and Non-Inferiority Trials. Parexel, PSI sponsored course notes: UK, 2000..
O'Brien PC, Fleming TR . A multiple testing procedure for clinical trials. Biometrics 1979; 35: 549–556.
Dolan P . Modeling valuations for EuroQol health states. Med Care 1997; 35: 1095–1108.
Acknowledgements
ARCTIC is funded by the Health Technology Assessment (HTA) programme and is included in the National Institute for Health Research (NIHR) Clinical Research Network Portfolio. The views and opinions expressed there in are those of the authors and do not necessarily reflect those of HTA, NIHR or the NHS. We would like to thank all patients and hospital staff who contributed to this study. In addition, we acknowledge the invaluable support provided by the independent DMC and TSC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Professor Hillmen received research funding and speakers' fees from Roche Products Limited. Dr Rawstron reports personal fees from Roche Products Limited. Dr Munir reports personal fees from Roche Products Limited. The remaining authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article

Cite this article
Howard, D., Munir, T., McParland, L. et al. Results of the randomized phase IIB ARCTIC trial of low-dose rituximab in previously untreated CLL. Leukemia 31, 2416–2425 (2017). https://doi.org/10.1038/leu.2017.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.96
This article is cited by
-
Combined analysis of IGHV mutations, telomere length and CD49d identifies long-term progression-free survivors in TP53 wild-type CLL treated with FCR-based therapies
Leukemia (2022)
-
Whole-genome sequencing of chronic lymphocytic leukemia identifies subgroups with distinct biological and clinical features
Nature Genetics (2022)
-
Methods and role of minimal residual disease after stem cell transplantation
Bone Marrow Transplantation (2019)
-
Telomere length predicts for outcome to FCR chemotherapy in CLL
Leukemia (2019)
-
An increased fraction of circulating miR-363 and miR-16 is particle bound in patients with chronic lymphocytic leukaemia as compared to normal subjects
BMC Research Notes (2018)
