
Abstract
The use of darbepoetin alfa to treat anemia in patients with lower-risk myelodysplastic syndromes (MDS) was evaluated in a phase 3 trial. Eligible patients had low/intermediate-1 risk MDS, hemoglobin ⩽10 g/dl, low transfusion burden and serum erythropoietin (EPO) ⩽500 mU/ml. Patients were randomized 2:1 to receive 24 weeks of subcutaneous darbepoetin alfa 500 μg or placebo every 3 weeks (Q3W), followed by 48 weeks of open-label darbepoetin alfa. A total of 147 patients were randomized, with median hemoglobin of 9.3 (Q1:8.8, Q3:9.7) g/dl and median baseline serum EPO of 69 (Q1:36, Q3:158) mU/ml. Transfusion incidence from weeks 5–24 was significantly lower with darbepoetin alfa versus placebo (36.1% (35/97) versus 59.2% (29/49), P=0.008) and erythroid response rates increased significantly with darbepoetin alfa (14.7% (11/75 evaluable) versus 0% (0/35 evaluable), P=0.016). In the 48-week open-label period, dose frequency increased from Q3W to Q2W in 81% (102/126) of patients; this was associated with a higher hematologic improvement–erythroid response rate (34.7% (34/98)). Safety results were consistent with a previous darbepoetin alfa phase 2 MDS trial. In conclusion, 24 weeks of darbepoetin alfa Q3W significantly reduced transfusions and increased rates of erythroid response with no new safety signals in lower-risk MDS (registered as EudraCT#2009-016522-14 and NCT#01362140).
Similar content being viewed by others
Introduction
Patients with low or intermediate-1 (int-1) International Prognostic Scoring System (IPSS) risk myelodysplastic syndromes (MDS) have a median survival of 5.7 and 3.5 years, respectively, when treated with supportive care, that is, primarily red blood cell (RBC) transfusion support.1 The main concern for these patients is anemia, a hallmark of MDS which mostly defines the disease burden in low/int-1 risk MDS.2 Anemia symptoms have various manifestations and alter patients’ quality of life. Accordingly, many patients receive RBC transfusions, which can lead to iron overload, cardiac and liver morbidity and mortality, and are associated with worse prognosis.2 Prevention or reduction of transfusions is therefore the main goal of clinical care in lower-risk MDS.
Erythropoiesis-stimulating agents (ESAs), such as epoetin alfa or darbepoetin alfa, are recommended in clinical guidelines to treat anemia in patients with lower-risk MDS, who generally have low transfusion burden.3, 4, 5 These recommendations are based on several phase 2 and 3 trials,6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24 including a phase 3 trial of daily erythropoietin (EPO) with or without granulocyte colony-stimulating factor with supportive care versus supportive care alone.8 In this trial there was an increased erythroid response rate with EPO treatment per International Working Group (IWG) 2000 criteria;25 response to EPO was associated with improved survival.
This is in line with other reports that patients responding to ESAs have a more favorable prognosis, with lower probability of progressing to acute myeloid leukemia (AML) and longer overall survival.8, 17, 26, 27 Main predictors of response are transfusion burden and endogenous EPO levels.28 In a recent meta-analysis of 55 trials, transfusion independence was associated with decreased mortality (hazard ratio of 0.41; 95% confidence interval (CI): 0.29–0.56).29 However, improved survival with ESAs has not been demonstrated in prospective randomized trials. Despite all the clinical trials, ESAs are not approved globally for use in MDS (approved in the European Union, Japan and Turkey). This lack of approval, mainly due to the paucity of placebo-controlled data, could limit access and impact patient care.
Here we describe the results of the largest prospective, phase 3, randomized, placebo-controlled multicenter trial to date of ESA treatment in lower-risk MDS, evaluating efficacy and safety of subcutaneous darbepoetin alfa in patients with IPSS low/int-1 risk MDS, anemia and low transfusion burden.
Patients and methods
Patients and study design
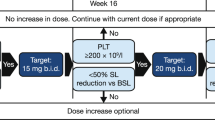
This was a phase 3, randomized, double-blind, placebo-controlled study of darbepoetin alfa in ESA-naive patients with low/int-1 risk MDS and anemia (Figure 1, https://eudract.ema.europa.eu/index.html, EudraCT#2009-016522-14 and https://clinicaltrials.gov/, NCT#01362140). The study was approved by the relevant institutional review boards/ethics committees and conducted in accordance with the Helsinki Declaration. Each patient signed an approved informed consent form.

Study design.
Eligible patients, enrolled in nine European countries from December 2011 to August 2014, had MDS per World Health Organization 2008 criteria with IPSS low/int-1 risk (determined locally), anemia (hemoglobin ⩽10 g/dl), endogenous serum EPO ⩽500 mU/ml, low transfusion burden (<4 RBC transfusion units in each of two consecutive 8-week periods before randomization), no previous treatment with ESAs or disease-modifying treatments and no bone marrow fibrosis. Patients were stratified by screening IPSS risk (low/int-1) for 2:1 randomization to 24 weeks of darbepoetin alfa or placebo followed by 48 weeks of open-label darbepoetin alfa for all patients and ongoing long-term follow-up for up to 3 years on study (although there is no investigational product (IP) during follow-up, any treatment, including commercially available ESAs, was allowed, per investigator). Transfusion endpoints were assessed from week 5 onward to allow adequate time for the effects of darbepoetin alfa to be observed.
Endpoints
Based on a phase 2 trial,6 an estimated 40% hematologic improvement–erythroid response (HI-E) per IWG 2006 criteria was used to calculate the sample size, with HI-E defined as ⩾1.5 g/dl increase from baseline in hemoglobin with a mean rise of ⩾1.5 g/dl for 8 weeks.30 During routine monitoring and review of blinded data by Amgen in 2013 (with ~1/2 expected trial patients), the cumulative HI-E response rate was lower than expected, making it less likely to detect a difference. Therefore, before unblinding and while enrollment was ongoing, Amgen, with input from regulatory bodies, recommended that transfusion incidence from weeks 5–24 become the primary endpoint and HI-E a secondary endpoint. This switch of endpoints was accepted by regulatory bodies in all countries except for Germany, where the Federal Institute for Drugs and Medical Devices (Bundesinstitut für Arzneimittel und Medizinprodukte) proceeded with the original endpoints (HI-E as primary, transfusion incidence as secondary).
Randomization and blinding
Eligible patients were randomized to darbepoetin alfa or placebo in a 2:1 ratio via an interactive voice response system, based on a schedule generated by Amgen before study start. Randomization was stratified by screening IPSS risk (low versus int-1). During the double-blind treatment period, the patient, site personnel and Amgen staff were blinded to treatment assignments. Darbepoetin alfa was provided as a clear, colorless, sterile, preservative-free, polysorbate solution in single-dose vials containing 100, 200, 300 or 500 μg/ml of darbepoetin alfa. Placebo was provided in identical containers.
Dosing
During the 24-week double-blind treatment period, patients received darbepoetin alfa 500 μg or matched placebo subcutaneously once every 3 weeks (Q3W) from day 1/week 1 to week 22. During the active treatment period (week 25 on), all patients could receive darbepoetin alfa 500 μg Q3W, except those who had dose reductions, who continued with the last dose assigned. Dose escalation was permitted only from weeks 31–71. For patients with a hemoglobin increase of <1.5 g/dl (relative to week 1 for patients randomized to darbepoetin alfa and week 25 for patients randomized to placebo) in the absence of RBC transfusions in the previous 28 days, the dose was escalated from 500 μg Q3W to 500 μg once every 2 weeks (Q2W) for the remainder of the active treatment period. Once switched to Q2W dosing, patients did not switch back to Q3W dosing (even if the dose was later reduced) to minimize visit schedule changes.
If a patient’s hemoglobin reached >12 g/dl, the dose was withheld until hemoglobin decreased to ⩽11.0 g/dl; then treatment was restarted at a reduced dose. If hemoglobin increased by >1.5 g/dl in 21 days for Q3W dosing or >1.0 g/dl in 14 days for Q2W dosing, in the absence of RBC transfusions, the dose was reduced. IP was discontinued and the patient entered long-term follow-up if >3 dose reductions were needed. Patients received whole blood or packed RBC transfusions when hemoglobin decreased to ⩽8.0 g/dl or as clinically indicated (for example, symptomatic).
Assessments
At each visit in the 24-week blinded and 48-week active treatment periods, patients were assessed for adverse events and thrombovascular events, and complete blood count and hemoglobin values were obtained. Ongoing independent central review was performed when local assessments found disease progression to AML or myelofibrosis or reticulin fibrosis. At weeks 1, 13, 25, 31, 42/43, 54/55 and 72/73, quality-of-life (QoL) assessments, Functional Assessment of Chronic Illness Therapy-Fatigue and EuroQol five dimensions visual analog scale were performed. Antibodies against darbepoetin alfa and EPO were assessed at weeks 1 and 25.
For exploratory independent expert review, blinded data were provided for hemoglobin (central and local), IP dose and schedule, any discontinuations or withdrawals for safety and transfusions. The three external experts reviewed each patient for clinical erythroid response, which was based on IWG 2006 criteria and also included changes in response duration if IP was discontinued for hemoglobin >12 g/dl, response to Q2W dosing during the active-treatment period, clinically meaningful transfusion threshold changes during the study (versus screening) and that baseline hemoglobin elevated by recent transfusions would affect response assessment.
Statistical analysis
The primary endpoint, RBC transfusion incidence weeks 5–24, was analyzed using a χ2-test for differences in proportions, with sensitivity analyses using the Cochran-Mantel–Haenszel χ2-test stratified by IPSS risk. For HI-E rates, a Cochran-Mantel–Haenszel χ2-test stratified by IPSS risk was used, with a sensitivity analysis using the χ2-test.
Endpoints were hierarchically tested to maintain the overall significance of 0.05. Hypothesis testing used a two-sided significance of 0.05. For all other endpoints, statistical testing was descriptive and no further adjustments were made for multiplicity. The planned sample size was set to 141; assumptions included 85% power, two-sided α of 5%, dropout rate of 5% and 135 evaluable patients (software: nQuery Advisor version 7.0).
Results
Patient characteristics and disposition
Patients (N=147) were randomized 2:1 to receive darbepoetin alfa (n=98) or placebo (n=49); baseline demographic and disease characteristics were generally similar (Table 1). Double-blind period completion rates were darbepoetin alfa: 89% (87/98); placebo: 80% (39/49). At the end of the 48-week open-label period, 117 patients continued in long-term follow-up (Figure 2).

Patient disposition. aPrimary analysis sets. bSerum EPO⩽500 mU/ml locally but >500 mU/ml centrally, so patient was withdrawn. cFor adverse event (n=2), protocol (n=2), administrative (n=1) and other (n=1). dFor adverse event (n=2), consent withdrawn (n=1) and protocol (n=1). eColon adenocarcinoma (T1aN0M0) treated with polypectomy. fPer dosing algorithm (n=9), progression to AML (n=2, both prior darbepoetin alfa). gPer investigator (n=2), lack of response (n=2) and unknown (n=3).
Efficacy: transfusions, HI-E and QoL
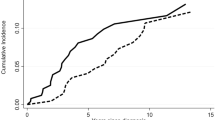
Transfusion incidence from weeks 5–24 was significantly reduced with darbepoetin alfa (odds ratio (95% CI) 0.38 (0.19–0.79), darbepoetin alfa: 36.1% (35/97), placebo: 59.2% (29/49), P=0.008) (Figure 3a). The mean (s.e.) numbers of transfusion episodes were darbepoetin alfa: 1.4 (0.2) and placebo: 2.7 (0.5) (P<0.001), and units transfused were darbepoetin alfa: 2.2 (0.4) and placebo: 4.1 (0.9) (P=0.038). Transfusion rates were less with lower baseline EPO for darbepoetin alfa (⩽100 mU/ml: 23%, >100 mU/ml: 57%, 95% CI non-overlapping) but not placebo. The proportion achieving HI-E per IWG 2006 criteria was significantly increased with darbepoetin alfa versus placebo in the 24-week double-blind period (darbepoetin alfa: 14.7% (95% CI: 7.6–24.7%) (11/75 evaluable), placebo: 0% (95% CI: 0-10%) (0/35 evaluable), P=0.016) (Figure 3b, unevaluable reasons in figure legend, response rate and disposition in Supplementary Figure S1). An erythroid response rate in favor of darbepoetin alfa was also found by post hoc exploratory independent blinded expert panel review (darbepoetin alfa: 23.6% (21/89), placebo: 4.2% (2/48)). All patients with HI-E (n=11) in the double-blind period had baseline serum EPO ⩽100 mU/ml, 4/11 had a dose withheld for hemoglobin >12 g/dl and 10/11 had no transfusions in the 16 weeks before randomization (none had transfusions in the 8 weeks before). For the 24-week period, the HI-E rate for each World Health Organization category was refractory anemia: 22% (2/9), refractory anemia with ringed sideroblasts: 12% (2/17), refractory cytopenias with multilineage dysplasia: 13% (6/45) and refractory anemia with excess blasts-1: 8% (1/13) (0% for other categories). A comparison of responses per IWG 2006 and historical IWG 2000 criteria is in Supplementary Table S1. When transfusion incidence and HI-E were examined by IPSS and revised IPSS (IPSS-R) risk (Supplementary Figure S2 and Supplementary Table S1), whereas changes by IPSS were not notably different, decreased transfusion requirements were associated with more favorable IPSS-R (P=0.005 for IPSS-R related to transfusion rates, P=0.56 for IPSS-R related to HI-E; both in the entire patient population).

Efficacy of darbepoetin alfa. (a) Transfusions assessed from week 5 onward to allow for the effects of darbepoetin alfa to be observed. (b) Erythroid response rates; reasons unevaluable for HI-E in the double-blind period (n=37) included transfusion in prior 28 days (n=33), no hemoglobin measurement within 14 days of first dose (n=3), did not receive darbepoetin alfa (n=1).
In the 48-week open-label period, the transfusion rate was 50.8% (64/126) and the HI-E rate was 34.7% (34/98) overall, 28.9% (11/38) for prior placebo and 26.4% (23/87) for prior darbepoetin alfa (10 of the prior darbepoetin alfa had HI-E in the 24-week portion). Of the 34 patients with HI-E, 6/34 (18%) had transfusions in the 16 weeks before randomization, 30/34 (88%) received Q2W dosing at some point, 30/34 (88%) had baseline serum EPO⩽100 mU/ml and 26/34 (76%) had doses withheld. For the 48-week period, the HI-E rate for each World Health Organization category was refractory anemia: 35% (7/20), refractory anemia with ringed sideroblasts: 15% (3/20), refractory cytopenias with multilineage dysplasia: 25% (13/53), del 5q: 23% (3/13), refractory anemia with excess blasts-1: 41% (7/17), MDS-U: 50% (1/2) and unknown: 0% (0/1). The mean (s.e.) duration of response was 235 (21) days, including both the 24-week double-blind and 48-week open-label treatment periods; 21/34 patients with HI-E were still responding at last observation.
For QoL assessments, for the 24-week double-blind period, the mean (s.d.) change from baseline with the EuroQol five dimensions visual analog scale did not differ by treatment (darbepoetin alfa (n=81): 2.1 (13.1) points, placebo (n=39): 0.8 (15.7) points). No difference was seen in rates of clinically meaningful (⩾3-point) improvement in Functional Assessment of Chronic Illness Therapy-Fatigue subscale score (darbepoetin alfa (n=85): 35.6% (95% CI: 25.7–46.4%), placebo (n=39): 31.0% (95% CI: 17.6–47.1%)). Further analyses of QoL data for the 24-week double-blind period by HI-E status, IPSS (low versus int-1), transfusion history (present/absent), hemoglobin response and stratification by median baseline QoL did not yield any significant differences. Likewise, for the subsequent 48-week open-label period, similar results were seen for prior darbepoetin alfa and prior placebo for EuroQol five dimensions visual analog scale and Functional Assessment of Chronic Illness Therapy-Fatigue.
Exposure
In the 24-week double-blind period, dose was reduced due to a rapid rise in hemoglobin (>1.5 g/dl in 3 weeks) in 18 (18%) darbepoetin alfa patients at a median (min–max) hemoglobin of 10.6 (8.4–12) g/dl (Table 2). Eleven (11%) darbepoetin alfa patients had a dose withheld for hemoglobin >12 g/dl; 4 of these 11 met HI-E criteria. In comparison, no placebo patients had the IP dose reduced or withheld.
In the 48-week open-label period, the median average dose administered was 500 μg. Fifty patients (40%) had a dose reduced a total of 85 times at a median (min–max) hemoglobin of 10.6 (8.5–11.9) g/dl. Dose was withheld for hemoglobin >12 g/dl in 36 (29%) patients. Dose frequency was increased from Q3W to Q2W in 81% (102/126) of patients.
Safety
During the double-blind treatment period, most adverse events were grade 1–2 in severity (Table 3). Adverse events leading to IP discontinuation were reported in two placebo-treated patients (grade 3 pulmonary arterial hypertension, grade 3 renal failure) and three darbepoetin alfa-treated patients (grade 3 pulmonary thrombosis, grade 3 thrombocytopenia, grade 1 increased blast cell count). There was no particular pattern for cardiovascular adverse events, thrombovascular adverse events, or adverse events of grade ⩾4 severity (Supplementary Tables S2, S3, and S4 respectively).
In the double-blind period, serious adverse events were reported in 11 darbepoetin alfa-treated patients (11.2%) and eight placebo-treated patients (16.7%) (Supplementary Table S5). The three fatal adverse events were hemorrhagic proctitis in the darbepoetin alfa group and one case each of cardiac failure and cerebral hemorrhage in the placebo group. The most frequently reported adverse events were patient-reported fatigue (darbepoetin alfa: 17.3%, placebo: 8.3%), asthenia (darbepoetin alfa: 12.2%, placebo: 10.4%) and exertional dyspnea (darbepoetin alfa: 6.1%, placebo: 10.4%) (Supplementary Table S6).
The incidence of disease progression to AML was similar in the darbepoetin alfa and placebo groups (2.1% versus 2.2%) (Supplementary Table S7). All AML cases were confirmed by central pathology; per central review, two patients who developed AML were refractory anemia with excess blasts-2 at baseline, not refractory anemia with excess blasts-1 as determined locally. Per protocol, these patients discontinued IP after AML diagnosis. One darbepoetin alfa-treated patient was diagnosed with stage 1A colon adenocarcinoma (T1aN0M0) 4 months after initiating treatment and was treated with polypectomy. No neutralizing antibodies to darbepoetin alfa or EPO were detected in those with post-baseline results (darbepoetin alfa N=91, placebo N=43). Regarding neutrophils and platelets, no significant differences from baseline or between groups were observed.
Adverse events reported in the 48-week open-label period were generally similar to those observed in the 24-week treatment period and similar between prior placebo and prior darbepoetin alfa groups (Table 3 and Supplementary Tables S2–S7). IP discontinuation due to adverse events occurred in three prior darbepoetin alfa patients (lung disorder, tetany, MDS progression, renal disorder and deep vein thrombosis) and three prior placebo patients (pulmonary embolism, anemia and delirium). Deaths included one of the AML cases (prior darbepoetin alfa) and pneumonitis (prior placebo). There were no neutralizing antibodies found to either darbepoetin alfa or EPO in this period (number of patients with post-baseline results: prior darbepoetin alfa N=80, prior placebo N=35).
Discussion
In this first phase 3, randomized, double-blind, placebo-controlled prospective trial of subcutaneous darbepoetin alfa in patients with IPSS low/int-1 risk MDS and anemia, darbepoetin alfa Q3W for 24 weeks significantly reduced transfusion incidence and increased rates of erythroid response per IWG 2006 criteria. These results are particularly notable as, in daily practice, the aim in managing patients with lower-risk MDS is to achieve transfusion independence, which is associated with improved survival.8, 17, 26, 27, 29 Safety findings were consistent with the known darbepoetin alfa safety profile and the phase 2 trial,6 with no new safety signals identified and an AML incidence of 2% in each group during the double-blind treatment period. Of note, we did not observe a significant increase in thromboembolic adverse events, which is consistent with previously reported results.24 The safety profile in the 48-week open-label period was similar for patients who had previously received placebo and those previously receiving darbepoetin alfa, indicating that longer darbepoetin alfa exposure was not associated with increased risk of adverse events. Although no significant changes were seen in QoL, the study was not powered for these analyses and the relatively small number of responders made it difficult to detect improved QoL. Further, that all patients, independent of treatment, received transfusions as needed could have made it harder to detect a difference in QoL.
Data from the 48-week open-label darbepoetin alfa period show an increased HI-E rate compared with the 24-week double-blind period; this may reflect that 81% of patients increased their dose frequency from Q3W to Q2W in the 48-week open-label period (increased dose frequency only allowed weeks 31–71). This HI-E rate is comparable to that of a phase 3 placebo-controlled study of epoetin-alfa in patients with low/int-1 MDS (31.8% versus 4.4% at 24 weeks); dosing in the epoetin-alfa study could be escalated after 8 weeks and adjusted weekly thereafter. The increased response rate may also reflect that prolonged treatment may be needed to obtain the full clinical benefit.31 These data indicate that a substantial number of patients receiving the 500 μg Q3W regimen may have been underdosed (as described by Fenaux and Adès32), accounting for the low response rate, and that Q2W dosing may offer more clinical benefit. In support of that, a recent systematic review of 10 darbepoetin alfa MDS studies found greater response rates in patients receiving doses equivalent to those in the open-label period of this trial.16 Further, the substantial number of patients (42%) with a history of RBC transfusions (albeit still low transfusion burden) may have influenced the outcomes of this trial.
The nature of the stringent IWG 2006 HI-E criteria (1.5 g/dl hemoglobin increase maintained for 8 weeks, even in patients with low transfusion burden) likely led to a lower than expected clinical benefit rate. The independent expert panel, which in addition to IWG 2006 criteria considered several variables before and during the study on a patient-by-patient basis, identified more patients with a clinical erythroid response as compared with the assessment strictly per the published version of the IWG 2006 criteria without additional clinical considerations. These data highlight important clinical factors not incorporated or reflected by current IWG criteria; these factors should be considered for any future revisions of IWG criteria.
Detecting erythroid response was further complicated by the trial design in several ways. First, hemoglobin was measured every 3 weeks, so in practice, patients had to maintain response over 9 weeks. Second, the darbepoetin alfa dose was held if hemoglobin rose to >12 g/dl and decreased if hemoglobin increased by >1.5 g/dl in 3 weeks (or 1 g/dl in 2 weeks for those dosed Q2W). This resulted in some patients having to reduce the darbepoetin alfa dose while still anemic, with dose reductions occurring with hemoglobin as low as 8.4 g/dl. Early dose reduction in patients with a rapid hemoglobin rise who were still anemic could have hampered the achievement of sustained hemoglobin responses and thus could have lowered the erythroid response rate. Third, as mentioned earlier, during the 48-week open-label period, 81% of patients changed from Q3W to Q2W dosing, indicating that optimal dosing was not achieved during the 24-week double-blind period. When erythroid response was measured as meeting IWG 2000 criteria at any point (that is, not necessarily for 8 weeks), higher response rates were seen (major response: 19%, minor response: 39%; Supplementary Table S1).
Our findings are in keeping with other ESA MDS trials, indicating that our results are likely generalizable to other patients with lower-risk MDS and anemia. Previously, in a phase 3 trial of EPO with supportive care versus supportive care alone, an increased erythroid response rate was observed with EPO, with no difference observed in survival or transformation to AML, with a median follow-up of 5.8 years.8 Retrospective analyses of Spanish and Greek MDS registries of patients with lower-risk MDS found that many patients responded to ESAs.27, 33 Further, ESA treatment in MDS was associated with improved survival and no increase in AML in a cohort study.26, 27 We also confirm the predictive value of IPSS-R risk with regard to transfusion response; thus, IPSS-R risk, in addition to baseline transfusion burden and endogenous EPO levels, can serve as an important marker to guide clinical care.
In conclusion, in this large phase 3, randomized, double-blind, placebo-controlled trial of darbepoetin alfa in IPSS low/int-1 risk MDS patients with anemia, subcutaneous darbepoetin alfa significantly reduced transfusions and increased rates of erythroid response compared with placebo with no new safety signals. Thus, darbepoetin alfa, at a dose of 500 μg given every 2 weeks, can be a meaningful treatment option for low/int-1 risk MDS patients suffering from anemia.
References
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997; 89: 2079–2088.
Platzbecker U, Hofbauer LC, Ehninger G, Holig K . The clinical, quality of life, and economic consequences of chronic anemia and transfusion support in patients with myelodysplastic syndromes. Leuk Res 2012; 36: 525–536.
Malcovati L, Hellstrom-Lindberg E, Bowen D, Ades L, Cermak J, Del Canizo C et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood 2013; 122: 2943–2964.
National Comprehensive Cancer Network I. Myelodysplastic Syndromes. 2016 (cited 3 March 2016); Available from https://www.nccn.org/professionals/physician_gls/f_guidelines.asp.
Onkologie DGfHuM. German S3 Leitlinie Guidelines. 2016 (cited 11 October 2016); Available from https://www.onkopedia.com/de/onkopedia/guidelines/myelodysplastische-syndrome-mds/@@view/html/index.html#ID0EIIBG.
Gabrilove J, Paquette R, Lyons RM, Mushtaq C, Sekeres MA, Tomita D et al. Phase 2, single-arm trial to evaluate the effectiveness of darbepoetin alfa for correcting anaemia in patients with myelodysplastic syndromes. Br J Haematol 2008; 142: 379–393.
Gotlib J, Lavori P, Quesada S, Stein RS, Shahnia S, Greenberg PL . A Phase II intra-patient dose-escalation trial of weight-based darbepoetin alfa with or without granulocyte-colony stimulating factor in myelodysplastic syndromes. Am J Hematol 2009; 84: 15–20.
Greenberg PL, Sun Z, Miller KB, Bennett JM, Tallman MS, Dewald G et al. Treatment of myelodysplastic syndrome patients with erythropoietin with or without granulocyte colony-stimulating factor: results of a prospective randomized phase 3 trial by the Eastern Cooperative Oncology Group (E1996). Blood 2009; 114: 2393–2400.
Jang JH, Harada H, Shibayama H, Shimazaki R, Kim HJ, Sawada K et al. A randomized controlled trial comparing darbepoetin alfa doses in red blood cell transfusion-dependent patients with low- or intermediate-1 risk myelodysplastic syndromes. Int J Hematol 2015; 102: 401–412.
Kelaidi C, Beyne-Rauzy O, Braun T, Sapena R, Cougoul P, Ades L et al. High response rate and improved exercise capacity and quality of life with a new regimen of darbepoetin alfa with or without filgrastim in lower-risk myelodysplastic syndromes: a phase II study by the GFM. Ann Hematol 2013; 92: 621–631.
Mannone L, Gardin C, Quarre MC, Bernard JF, Vassilieff D, Ades L et al. High-dose darbepoetin alpha in the treatment of anaemia of lower risk myelodysplastic syndrome results of a phase II study. Br J Haematol 2006; 133: 513–519.
Moyo V, Lefebvre P, Duh MS, Yektashenas B, Mundle S . Erythropoiesis-stimulating agents in the treatment of anemia in myelodysplastic syndromes: a meta-analysis. Ann Hematol 2008; 87: 527–536.
Musto P, Lanza F, Balleari E, Grossi A, Falcone A, Sanpaolo G et al. Darbepoetin alpha for the treatment of anaemia in low-intermediate risk myelodysplastic syndromes. Br J Haematol 2005; 128: 204–209.
Nilsson-Ehle H, Birgegard G, Samuelsson J, Antunovic P, Astermark J, Garelius H et al. Quality of life, physical function and MRI T2* in elderly low-risk MDS patients treated to a haemoglobin level of ⩾120 g/L with darbepoetin alfa +/- filgrastim or erythrocyte transfusions. Eur J Haematol 2011; 87: 244–252.
Oliva EN, Nobile F, Alimena G, Specchia G, Danova M, Rovati B et al. Darbepoetin alfa for the treatment of anemia associated with myelodysplastic syndromes: efficacy and quality of life. Leuk Lymph 2010; 51: 1007–1014.
Park S, Fenaux P, Greenberg P, Mehta B, Callaghan F, Kim C et al. Efficacy and safety of darbepoetin alpha in patients with myelodysplastic syndromes: a systematic review and meta-analysis. Br J Haematol 2016; 174: 730–747.
Park S, Grabar S, Kelaidi C, Beyne-Rauzy O, Picard F, Bardet V et al. Predictive factors of response and survival in myelodysplastic syndrome treated with erythropoietin and G-CSF: the GFM experience. Blood 2008; 111: 574–582.
Patton JF, Sullivan T, Mun Y, Reeves T, Rossi G, Wallace JF . A retrospective cohort study to assess the impact of therapeutic substitution of darbepoetin alfa for epoetin alfa in anemic patients with myelodysplastic syndrome. J Support Oncol 2005; 3: 419–426.
Stasi R, Abruzzese E, Lanzetta G, Terzoli E, Amadori S . Darbepoetin alfa for the treatment of anemic patients with low- and intermediate-1-risk myelodysplastic syndromes. Ann Oncol 2005; 16: 1921–1927.
Villegas A, Arrizabalaga B, Fernandez-Lago C, Castro M, Mayans JR, Gonzalez-Porras JR et al. Darbepoetin alfa for anemia in patients with low or intermediate-1 risk myelodysplastic syndromes and positive predictive factors of response. Curr Med Res Opin 2011; 27: 951–960.
Hellström-Lindberg E, van de Loosdrecht A . Erythropoiesis stimulating agents and other growth factors in low-risk MDS. Best Pract Clin Haematol 2013; 26: 401–410.
Jädersten M, Montgomery SM, Dybedal I, Porwit-MacDonald A, Hellstrom-Lindberg E . Long-term outcome of treatment of anemia in MDS with erythropoietin and G-CSF. Blood 2005; 106: 803–811.
Rigolin GM, Porta MD, Ciccone M, Bugli AM, Bragotti LZ, Mauro E et al. In patients with myelodysplastic syndromes response to rHuEPO and G-CSF treatment is related to an increase of cytogenetically normal CD34 cells. Br J Haematol 2004; 126: 501–507.
Smith SW, Sato M, Gore SD, Baer MR, Ke X, McNally D et al. Erythropoiesis-stimulating agents are not associated with increased risk of thrombosis in patients with myelodysplastic syndromes. Haematologica 2012; 97: 15–20.
Cheson BD, Bennett JM, Kantarjian H, Pinto A, Schiffer CA, Nimer SD et al. Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood 2000; 96: 3671–3674.
Jädersten M, Malcovati L, Dybedal I, Della Porta MG, Invernizzi R, Montgomery SM et al. Erythropoietin and granulocyte-colony stimulating factor treatment associated with improved survival in myelodysplastic syndrome. J Clin Oncol 2008; 26: 3607–3613.
Symeonidis A, Zikos P, Galanopoulos A, Kotsianidis I, Kouraklis A, Protopapa M et al. Response to treatment with erythropoietin in patients with MDS highly predicts low risk of evolution to AML and longer survival. Leuk Res 2011; 35: S127–S128.
Hellström-Lindberg E, Gulbrandsen N, Lindberg G, Ahlgren T, Dahl IM, Dybedal I et al. A validated decision model for treating the anaemia of myelodysplastic syndromes with erythropoietin + granulocyte colony-stimulating factor: significant effects on quality of life. Br J Haematol 2003; 120: 1037–1046.
Harnan S, Ren S, Gomersall T, Everson-Hock ES, Sutton A, Dhanasiri S et al. Association between transfusion status and overall survival in patients with myelodysplastic syndromes: a systematic literature review and meta-analysis. Acta Haematol 2016; 136: 23–42.
Cheson BD, Greenberg PL, Bennett JM, Lowenberg B, Wijermans PW, Nimer SD et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006; 108: 419–425.
Terpos E, Mougiou A, Kouraklis A, Chatzivassili A, Michalis E, Giannakoulas N et al. Prolonged administration of erythropoietin increases erythroid response rate in myelodysplastic syndromes: a phase II trial in 281 patients. Br J Haematol 2002; 118: 174–180.
Fenaux P, Adès L . How we treat lower-risk myelodysplastic syndromes. Blood 2013; 121: 4280–4286.
Diez Campelo M, Lorenzo JI, Benlloch L, Lopez-Pavia M, Such E, Bernal T et al. Spanish registry of erythropoietic stimulating agents study: The largest retrospective study of ESAs for the treatment of anemia in lower risk MDS patients. Leuk Res 2015; 39: S105–S106.
Acknowledgements
All authors had access to the complete clinical dataset. Susanna Mac of Amgen Inc. provided medical writing support. Amgen Inc. ran and funded this study and all analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
UP: honoraria from Amgen Inc., Celgene, Janssen and Novartis. AS: research grants from Amgen Inc. ENO: honoraria from Celgene, Amgen Inc., La Jolla and speakers’ bureau for Novartis, Celgene. JSG: honoraria from Amgen Inc. MD: honoraria from Amgen Inc. JM: research grants from Amgen Inc. BS: none. SB, BM and JF: employees and stockholders of Amgen Inc. EG: Novartis employee, former Amgen Inc. employee.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Platzbecker, U., Symeonidis, A., Oliva, E. et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia 31, 1944–1950 (2017). https://doi.org/10.1038/leu.2017.192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2017.192
This article is cited by
-
Iron overload induces dysplastic erythropoiesis and features of myelodysplasia in Nrf2-deficient mice
Leukemia (2024)
-
Treatment of Myelodysplastic Syndromes for Older Patients: Current State of Science, Challenges, and Opportunities
Current Hematologic Malignancy Reports (2024)
-
Current Therapeutic Landscape in Lower Risk Myelodysplastic Syndromes
Current Treatment Options in Oncology (2023)
-
Myelodysplastic syndromes
Nature Reviews Disease Primers (2022)
-
New Approaches to Myelodysplastic Syndrome Treatment
Current Treatment Options in Oncology (2022)
