
Abstract
Recent studies suggest that a proportion of chronic myeloid leukemia (CML) patients in deep molecular remission can discontinue the tyrosine kinase inhibitor (TKI) treatment without disease relapse. In this multi-center, prospective clinical trial (EURO-SKI, NCT01596114) we analyzed the function and phenotype of T and NK cells and their relation to successful TKI cessation. Lymphocyte subclasses were measured from 100 imatinib-treated patients at baseline and 1 month after the discontinuation, and functional characterization of NK and T cells was done from 45 patients. The proportion of NK cells was associated with the molecular relapse-free survival as patients with higher than median NK-cell percentage at the time of drug discontinuation had better probability to stay in remission. Similar association was not found with T or B cells or their subsets. In non-relapsing patients the NK-cell phenotype was mature, whereas patients with more naïve CD56bright NK cells had decreased relapse-free survival. In addition, the TNF-α/IFN-γ cytokine secretion by NK cells correlated with the successful drug discontinuation. Our results highlight the role of NK cells in sustaining remission and strengthen the status of CML as an immunogenic tumor warranting novel clinical trials with immunomodulating agents.
Similar content being viewed by others

Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative cancer that seeds from a translocation (9;22) in the hematopoietic stem cell resulting in constitutively active BCR-ABL1 oncokinase. The inhibition of BCR-ABL1 with tyrosine kinase inhibitors (TKIs) has revolutionized the prognosis of CML.1, 2, 3, 4 The first TKI developed for the treatment of CML (imatinib) has now been in use for 15 years. However, TKIs are not considered to be curative as the majority of patients still have residual disease left after years on treatment.5 Even though therapy responses to TKIs are generally very good, the life-long medication creates physiological, mental and economical burden.6 In addition, the prevalence of CML is increasing due to the improved treatment results.7 Therefore, there is a significant need to find novel treatment strategies aiming for cure. Recent reports suggest that approximately 40% of CML patients who have achieved optimal therapy response (deep molecular remission) can discontinue imatinib treatment without recurrence of detectable BCR-ABL1 transcripts.8, 9, 10 Similarly dasatinib discontinuation after sustained deep molecular response has shown to be successful in 50% of patients.11 However, with more sensitive DNA-based methods residual leukemic cells can still be detected in blood samples from these patients.9 To be able to cure CML we would either need to eradicate or alternatively regain the immune control of the remaining leukemic cells.
We set up an immunological study within the framework of the pan-European TKI stopping study (EURO-SKI) in order to understand whether the immune system has a role in the successful discontinuation of the TKI treatment. Here we show that a high proportion of mature NK cells is related to the successful imatinib discontinuation highlighting the importance of NK cells when considering future treatment strategies.
Materials and methods
Study patients and samples
The study was conducted by the Nordic CML study group (NCMLSG) as a substudy to the EURO-SKI clinical trial (NCT01596114). Altogether, 132 consecutive chronic phase CML patients who participated in the clinical EURO-SKI trial were recruited from the Nordic countries. Study participation was only based on the patient’s and treating physician’s willingness to take part in the immunology substudy protocol. Patients were treated with imatinib (n=107), dasatinib (n=15), or nilotinib (n=9) for at least three years prior to the study and had sustained deep molecular response (MR4) for at least 1 year (Supplementary Figure 1). After the TKI discontinuation, patients were closely followed with monthly RQ-PCR tests to monitor the level of residual leukemia cells. In the clinical study, relapse was defined as a loss of major molecular response (BCR-ABL1 transcripts >0.1% on the international scale (IS)). In the substudy, peripheral blood (PB) samples were collected before stopping TKI treatment and 1 and 6 months after. As the number of patients treated with second generation TKIs (dasatinib and nilotinib) was low, only results from imatinib-treated patients are presented (Supplementary Figure 1).
Basic NK-, B- and T-cell counts and proportions were analyzed with the flow cytometry in the accredited university hospitals. From a proportion of patients (n=45), who were willing to give extra 50 ml of blood for explorative analysis, a more detailed NK- and T-cell function and immune phenotype was studied (Supplementary Figure 1 and Supplementary Table 1). Thus, no biased patient selection was involved. All the functional analyses were performed centralized in the Helsinki laboratory (described in detail below).
All patients and healthy controls gave their written informed consent and the study was approved by local University Hospitals and conducted in accordance with the Declaration of Helsinki.
Immunophenotyping of NK-cells
Freshly isolated mononuclear cells (MNCs) were stained with CD45- CD3-, CD14-and CD19-, CD56-, CD16-, CD57, CD62L-, CD27- and CD45RA-antibodies and analysed with flowcytometry. For more detailed description see Supplementary Methods.
NK-cell cytokine secretion and degranulation assays
To study the NK-cell degranulation and cytokine secretion capability, fresh MNCs were stimulated with K562 or with PMA and Calcium Ionophore. In the cytokine secretion assay the intracellular tumor necrosis factor (TNF)-α and interferon (IFN)-γ secretion was measured and in the degranulation assay the expression CD107a/b was studied with flowcytometry (Supplementary Methods).
KIR genotyping
DNA was extracted from either fresh or thawed PB MNCs with NucleoSpin Tissue kit (#740952, Machery-Nagel, Duren, Germany) according to instructions or from fresh whole blood using magnetic beads and Chemagic MSM1 from PerkinElmer (Waltham, MA, USA). Patients were KIR-genotyped according to the manufacturer’s instructions by using Olerup SSP KIR Genotyping kit (Olerup SSP AB, Stockholm, Sweden). If any of the 2DL2, 2DL5, 3DS1, 2DS1, 2DS2, 2DS3 or 2DS5 genes was present, the haplotype was considered Bx. Accordingly, if none of these was present, the haplotype was AA.
T-cell phenotyping
Freshly isolated MNCs were stained with CD45-, CD3-, CD4-, CD45RA-, CD62L-, CD57- and –CD27-antibodies. To further study the T-cell phenotype, thawed MNCs were stained with CD3- CD4-, CD8-, CD45RA-, PD-1-, CTLA4-, CD28-, CCR7- and CXCR4-antibodies. For more detailed description see Supplementary Methods.
Activation of T-cells
Fresh MNCs were stimulated with anti-CD3 and co-stimulatory anti-CD28 and anti-CD49d and incubated for 6 h in the presence of GolgiStop. After intracellular staining the IFN-γ and TNF-α secretion was measured with flowcytometry. For more detailed description see Supplementary Methods.
Cytokine bead array
Plasma cytokine profiles (IL-2, IL-4, IL-6, IL-10, TNFα, IFNγ, IL-17A, IL-8, RANTES, MIG, MCP-1 and IP-10) were studied with cytokine bead array (BD Biosciences, San Jose, CA, USA) using Th1/Th2/Th17 cytokine and chemokine kits (BD Biosciences) according to the manufacture’s instructions.
Statistical analysis
Non-parametric Mann–Whitney test was used for comparison between two groups. One-way analysis of variance (ANOVA) was used for comparison between multiple groups, and Bonferroni’s post test was used to compare selected pairs if ANOVA gave significant P-value (P<0.05). Paired t-test was used to compare sequential samples from same patients.
Molecular recurrence-free survival was estimated by the Kaplan–Meier method and compared within groups by the log-rank test. For these longitudinal analyses, quantitative variables were dichotomized according to their median value, and for most promising variables also with receiver-operating characteristics (ROC) curves and the Youden index12 to optimize their cutoff points. Main results are provided with hazard ratio (HR) at 6 months and 95% confidence intervals (95% CI).
Correlation between different parameters was assessed with the use of non-parametric Spearman’s coefficient. All tests were two sided and P<0.05 was considered statistically significant. Analyses were performed with SAS v. 9 (SAS institute, Cary NJ, USA) and GraphPad Prism (GraphPad Software Inc., CA, USA).
Results
Increased NK-cell frequencies in non-relapsing patients before imatinib discontinuation
Although some in vitro studies have suggested that TKI therapy may have immunosuppressive effects,13, 14, 15 in the majority of patients, lymphocyte subsets were within normal range (Supplementary Figures 2 and 3). The median proportion of NK cells (CD3-CD56+/CD16+) among lymphocytes was increased in patients compared with controls (16 vs 11%, P=0.003), and also similar trend was observed in absolute NK cells counts (0.26 vs 0.21 × 109cells per l, P=0.05). In contrast, both absolute and relative B-cell numbers and absolute T-cell counts were decreased (Supplementary Figure 3). At 1 month after the TKI discontinuation only a slight increase in total WBC and monocyte counts was noted, but no marked changes among lymphocyte subsets such as T or NK cells (Supplementary Figures 2 and 3).
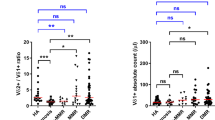
Analyses of the immune-cell subsets at the time of imatinib discontinuation demonstrated that the proportion of NK cells was associated with the molecular relapse-free survival. Patients whose relative proportion of NK cells was higher than median had increased molecular relapse-free survival compared with the patients with lower NK-cell proportion (73% vs 51% at 6 months, hazard ratio 2.17, P=0.02; Figure 1a and Table 1). Furthermore, when the optimal cutoff of the relative proportion of NK cells was determined according to ROC curves and Youden index, the difference in the molecular relapse-free survival between the high and low NK groups was even more pronounced (at 6 months 70% vs 43%, hazard ratio 2.35, P=0.009; Figure 1b and Supplementary Table 2).

The proportion and absolute count of NK cells at the time of imatinib discontinuation. (a) Molecular relapse-free survival of imatinib treated patients at baseline based on the median NK-cell proportion (in all patients median 16%, range 5–48%). Log-rank test was used to analyze the statistical significance between the two groups. Hazard ratio is reported in Table 1. (b) Patients were dichotomized to low- and high-NK-cell groups according to receiver-operating characteristics (ROC) curves and the Youden index to find more optimal cutoff for groups than the median value (AUROC 0.568, 95% CI: 0.4543–0.6818). On the basis of these analyses 11.3% was used as a cutoff. Hazard ratio is reported in the Supplementary Table 2. (c) Clinical characteristics of patients according to their TKI discontinuation success. Patients were divided in three groups: non-relapsing (able to maintain remission for at least 12 months, n=49), early relapsing (relapse before 6 months, n=34) and late relapsing patients (relapse after 6 months, n=17). ICF, immune cell function. (d) The relative number of NK cells at baseline in patient groups and healthy volunteers (n=48). Median early relapsing 12.8%, late relapsing 20%, non-relapsing 17.8% and healthy 11.5%. (e) Absolute NK-cell count at baseline in patient groups and healthy volunteers. Median early relapsing 0.19 × 109 cells per l, late-relapsing 0.31 × 109 cells per l, non-relapsing 0.25 × 109 cells per l and healthy 0.21 × 109 cells per l. Box-and-whisker plots present 5–95 percentiles. One-way ANOVA was used for comparison between multiple groups (exact P-value reported in the figure), and Bonferroni’s post test was used to compare selected pairs (only early-relapse group was compared with other groups to avoid multiple comparisons). Statistically significant differences between the groups are noted with asterisk (*P<0.05).
Similar association was not observed when the absolute or relative numbers of T (CD4+ and CD8+) and B cells or total monocyte, lymphocyte and leucocyte counts were analyzed.
As previous studies have shown that the majority of relapses occur within 6 months after imatinib discontinuation,8, 16 and as late relapses may have different biological background compared to relapses occurring immediately after TKI discontinuation, patients were divided into three different groups based on the discontinuation success (Figure 1c). Interestingly, patients in the early relapsing group had lower relative proportion of NK cells when compared with the non-relapsing patients (median 12.8% vs 17.1%, Figure 1d), whereas in the late relapsing group the median proportion of NK cells was at the same level as in the non-relapsing patients (20% of lymphocytes, Figure 1d). A similar trend was also observed with absolute NK-cell counts (median 0.19 × 109 cells per l vs 0.25 × 109 cells per l;, respectively; late relapsing 0.31 × 109 cells per l, Figure 1e). At 1 month time point after imatinib discontinuation, the differences between the groups still existed, and the early relapsing patients had significantly lower proportion of NK cells when compared with late or non-relapsing patients (Supplementary Figure 4).
Analysis of the clinical characteristics (gender, age, Sokal score, duration of TKI treatment and previous IFN-α treatment) showed no statistically significant correlation between the parameters and relative or absolute numbers of NK cells.
No association between KIR genes and successful imatinib discontinuation
As the function of NK cells is mediated with activating and inhibitory killer-cell immunoglobulin-like receptors (KIRs), we assessed the repertoire of KIR genes and AA and Bx haplotypes in individual patients by genotyping (n=43). No differences were noted in individual KIR gene frequencies or in the AA or Bx haplotype frequencies when non-relapsing, early and late relapsing groups were compared.
Increased proportion of CD3-CD56bright NK cells is related to rapid molecular relapse
To better understand the role of NK cells in successful treatment discontinuation, detailed immunophenotyping was performed. First, the proportion of CD56bright NK cells was assessed as they are regarded as immature NK cells, and in the maturation process they lose some of the CD56 receptors and turn into CD56dim NK cells.17 A trend for decreased molecular relapse-free survival was observed in patients who had higher than median number of CD56bright NK cells (at 6 months 52% vs 70%, hazard ratio 0.47, P=0.14; Figure 2a and Table 1). A more pronounced difference between the groups was observed when optimal cutoff for CD56bright low and high was determined (at 6 months 48% vs 79% respectively, hazard ratio 0.29, P=0.032; Figure 2b and Supplementary Table 2). In concord, both at the baseline and especially at the 1 month time point, both non- and early relapsing groups had higher proportion of more mature CD56dim NK cells than early relapsing group (Figure 2c and Supplementary Figure 5A).

NK-cell phenotype at the time of imatinib discontinuation. Ficoll isolated fresh PB MNCs were analyzed with multicolor flow cytometry. NK cells were defined as CD3-CD56+ lymphocytes. (a) Molecular relapse-free survival of imatinib treated patients at baseline based on the median proportion of CD56bright NK cells. Log-rank test was used to analyze the statistical significance between the two groups. Hazard ratio is reported in Table 1. (b) Patients were dichotomized to low and high CD56bright NK-cell groups according to ROC and the Youden index analyses (AUROC 0.6011; 95% CI: 0.4275–0.7748). 2.99% was used as a cutoff. Hazard ratio is reported in the Supplementary Table 2. (c) The proportion of CD56dim cells of CD56+ NK cells in early (n=13), late (n=8) and non-relapsing (n=19) patients (d) The proportion of CD57+ NK cells. (e) The proportion of CD16+ NK cells. In c–e Box-and-whisker plots present 5–95 percentiles. One-way ANOVA was used for comparison between multiple groups (exact P-value reported in the figure), and Bonferroni’s post test was used to compare selected pairs (only early relapse group was compared to other groups to avoid multiple comparisons). Statistically significant differences between the groups are marked with asterisk (*P<0.05, **P<0.01).
In addition to division for CD56bright and CD56dim cells, additional phenotypic markers were analyzed. In the early relapsing group, the proportion of CD57 expressing NK cells (a marker for highly differentiated NK cells18) was decreased at 1 month after imatinib discontinuation, and also the proportion of CD16-positive NK cells at baseline was significantly decreased (Figures 2c and d, and Supplementary Figures 5B and C). CD16 is a FC gamma receptor mediating antibody-dependent cell-mediated cytotoxicity (ADCC).19 However, when the expression of activating NK-cell receptors (NKG2C, NKp46 and NKG2D) affecting NK-cell function was analyzed, no significant differences in expression levels on CD3–CD56dim NK cells were observed between the non-relapsing and early relapsing groups (Supplementary Figure 6).
Adaptive NK cells are increased in non-relapsing patients
To further characterize the NK-cell phenotype in non-relapsing patients, recently identified markers (loss of FcɛRγ, PLZF, SYK, EAT-2) for adaptive NK cells20, 21 were analyzed. At the baseline, only a limited number of samples was available for the analysis (n=15), but non-relapsing patients appeared to have higher frequencies of adaptive-like CD3-CD56dim NK cells that had downregulated EAT-2 (2.8% vs 1.2% of lymphocytes, P<0.05, late relapsing 0.8%). At the 1-month time point after the imatinib discontinuation when bigger number of samples were available (n=39), results were more pronounced. Non-relapsing patients had significantly higher frequencies of NK cells that had down-regulated EAT-2 as compared with early relapsing patients (2.1% vs 0.6%, P<0.01, Figure 3a). In addition, when NK cells at 1 month were plotted according to the expression of the adaptive NK-cell marker (EAT-2–) and treatment-free time, positive correlation was observed (r=0.44, P=0.005, Figure 3b). However, with other markers (loss of FcɛRγ, PLZF, SYK) only trends were observed (Figure 3a).

Adaptive-like NK cells in treatment discontinuation. Frozen PB MNCs from 1-month time-point were analyzed with multicolor flow cytometry. (a) Adaptive-like NK cells based on the downregulation of EAT2, SYK, FCɛR1γ and PLZF expression at 1 month. EAT-2– in non-relapsing group 2.1% (n=19), early relapsing 0.5% (n=13), late relapsing 1.1% (n=7) of lymphocytes, **P<0.01. SYK– non-relapsing 3.3%, early relapsing 1.3%, late relapsing 1.3% of lymphocytes, P=NS. FcɛRγ– non-relapsing 5.9%, early relapsing 1.5%, late relapsing 3.5% of lymphocytes, P=0.18. PLZF– non-relapsing 3.7%, early relapsing 1.4%, late relapsing 5.0% of lymphocytes, P=0.11. Box-and-whisker plots present 5–95 percentiles. (b) The proportion of EAT2- adaptive-like NK cells at 1 month plotted according to the treatment-free remission time (n=39). For non-relapsing patients, the latest follow-up time is reported. Statistical significance was analyzed with non-parametric Mann–Whitney test (a) and with Spearman correlation (b).
CD56dimCD16–NK cells from non-relapsing imatinib treated patients have normalized cytokine secretion
To study the function of NK cells, PBMC from patients and controls were stimulated with K562 cells, a CML cell line devoid of MHC class I expression. Following target cell stimulation, degranulation and cytokine secretion was quantified on NK-cell subsets. For the major NK-cell subsets, CD3–CD56bright and CD3–CD56dim NK cells, no differences in the degranulation responses were observed between the different groups (data not shown). However, when studying baseline samples a higher frequency of CD3–CD56dimCD16– NK cells that excelled in production of TNF-α and IFN-γ upon stimulation was associated with increased molecular relapse-free survival both at 6 (85% vs 40%) and 12 months (69% vs 40%) compared to patients with lower TNF-α/IFN-γ secretion in the CD56dimCD16– NK-cell subset (hazard ratio at 6 months 5.57, P=0.008; Figure 4a and Table 1). When dichotomizing patients into high and low groups based on the ROC and Youden Index analysis, 81% of patients had molecular relapse-free survival at 6 months in the high group compared with 29% in the low group (hazard ratio 6.00, P=0.001; Figure 4b and Supplementary Table 2).

NK-cell activation and cytokine secretion at the time of imatinib discontinuation. MNCs collected at the baseline before imatinib discontinuation were stimulated with K562 cells for 6 hours at +37C. After, the cells were collected and stained for surface and intracellular markers and analyzed with flow cytometry. (a) Molecular relapse-free survival of imatinib treated patients at baseline based on the median proportion of CD16- NK cells secreting TNF-α/IFN-γ. Log-rank test was used to analyze the statistical significance between the two groups. Hazard ratio is reported in Table 1. (b) Patients were dichotomized to low and high TNF-α/IFN-γ secretion groups according to ROC and the Youden index analyses (AUROC 0.7175; 95% CI: 0.5580–0.8770). 14.05% was used as a cutoff. Hazard ratio is reported in the Supplementary Table 2. (c) CD16 downregulation in CD56dim NK cells after K562 stimulation. (d) The proportion of CD16- NK cells secreting TNF-α/IFN-γ in patient groups at baseline after K562 stimulation. Early relapse n=14, late relapse n= 6, non-relapse n=19, and healthy n=8. (c, d) Box-and-whisker plots present 5–95 percentiles. One-way ANOVA was used for comparison between multiple groups (exact P-value reported in the figure), and Bonferroni’s post test was used to compare selected pairs (only early relapse group was compared to other groups to avoid multiple comparisons). Statistically significant differences between the groups are marked with asterisk (*P<0.05, **P<0.01).
As CD16 can be shed upon target cell stimulation,22 further analysis showed that non-relapsing patients had more NK cells that had downregulated CD16 upon K562 stimulation (median fold change in non-relapsing patients 1.5 vs early relapsing 1.1, late relapse 1.4; Figure 4c) suggesting an increased activity of these cells. In accordance, in the non-relapsing group the response to K562 stimulation in activated CD56dimCD16– NK-cell subset was observed as an increased proportion of TNF-α/IFN-γ secreting cells (non-relapsing 18.5% vs early relapsing 12% of CD56dimCD16– NK cells, Figure 4d). In summary, these results can be interpreted as reflecting increased signaling among CD3–CD56dim NK cells upon target cell stimulation (shedding of CD16), leading to a higher proportion of cytokine-producing CD3–CD56dimCD16– NK cells in non-relapsing patients.
Cytokine secretion by CD4+ T cells correlates with the relative amount of NK cells
To study the role of T cells in imatinib discontinuation, T cells were phenotyped and stimulated with anti-CD3 antibodies to detect a Th1 type response (TNF-α/IFN-γ secretion). In the early-relapsing group of patients, there was a trend that CD8+ T cells were more immature (increased proportion of CD45RA+CD62L+ cells) compared with the non relapsing group (Figure 5a). No clear differences were observed in the memory phenotype of CD4+ T cells or in the amount of regulatory T cells. However, when the expression of PD-1 activating antigen was analyzed on the CD4+ T cells, the non-relapsing patient group had more PD-1 expressing central memory CD4+ T cells compared with the early-relapsing group (Figure 5b) suggesting recent activation.

T-cell immunophenotype and cytokine secretion analyzed with flow cytometry. (a) The proportion of naïve CD8+ CD45RA+CD62L+ T cells in early-relapsing patients (20.6%, n=13), late-relapsing patients (18.9%, n=8) and non-relapsing patients (12.5%, n=17). (b) PD-1 expressing central memory CD4+ T cells (CCR7+CD45RA-) in early relapsing (16.5%, n=9), late relapsing (16.0%, n=8) and non-relapsing patients at baseline (21.7% (n=12). (a, b) Box-and-whisker plots present 5–95 percentiles. One-way ANOVA was used for comparison between multiple groups (exact P-value reported in the figure), and Bonferroni’s post test was used to compare selected pairs (only early relapse group was compared with other groups to avoid multiple comparisons). Statistically significant differences between the groups are marked with asterisk (*P<0.05). (c) Correlation between TNF-α/IFN-γ secretion of CD4+ T cells with NK-cell proportion in non-relapsing patients at baseline. (d) Correlation between TNF-α/IFN-γ secretion of CD4+ T cells and NK-cell proportion in relapsing patients (early and late) at baseline. Correlations were analyzed with the non-parametric Spearman test.
No significant differences were observed in the Th1 type cytokine secretion between the groups. Interestingly though, there was a correlation between the CD4+ T-cell responses (TNF-α/IFN-γ secretion) and the relative number of NK cells. In the non-relapsing group, only tendency towards positive correlation was observed (r=0.43, P=0.07, Figure 5c), but in both relapsing groups negative correlation was found (r=−0.46, P<0.05, Figure 5d) suggesting that in relapsing patients NK cells may not suffice to activate CD4+ T cells to mount Th1 type responses.
As plasma cytokine milieu is important in controlling both the T- and NK-cell function, we also determined the plasma cytokine levels with bead arrays (IL-2, IL-4, IL-6, IL-10, TNF, IFN-γ, IL-17A, IL-8, RANTES, MIG, MCP-1, and IP-10). Only IL-10 was slightly increased in the non-relapsing patients (7 vs 5.8 pg/ml n=8, P=0.07, late relapsing 6 pg/ml), and no additional differences in the amounts of other cytokines was observed between the groups. Although IL-10 may have immunosuppressive functions, there are indications that IL-10 could also stimulate the immune system as it has been shown that IL-10 can act as a chemoattractant for NK cells.23
Discussion
Despite the success of TKIs, patients still suffer from long-term adverse effects, which can markedly affect the quality of life.24 In addition, the significant drug costs cause economical burden. Although the need for daily TKI therapy has earlier been considered to be life-long, recent clinical trials have suggested that at least a proportion of patients are able to discontinue the therapy and stay in remission. However, the mechanisms behind this are mostly unknown. As it has already been debated that TKI cessation may become a clinical practice in CML,25, 26 the understanding of biological basis for successful discontinuation is of utmost importance.
In the thus far largest CML TKI discontinuation trial (EURO-SKI clinical trial), we were able to thoroughly investigate the role of the immune system in sustained remission after treatment cessation. We found that the increased molecular relapse-free survival was associated with a higher proportion of NK cells. In addition, NK cells secreted TNF-α/IFN-γ cytokines, which are able to activate T cells. Accordingly, we noted that in non-relapsing patients the number of NK cells was associated with a Th1 type response of CD4+ T cells, which also may contribute to the antitumor immunity. Therefore, we hypothesize that an increased amount of mature NK cells may be capable of both directly killing the tumor cells and potentiating adaptive immune responses against leukemia, thereby maintaining remission after imatinib discontinuation (Supplementary Figure 7). It should be, however, noted, that other factors such as the Sokal score, IFN-α exposure or the duration of the treatment may have a role in successful TKI stopping,8 but according to our data the proportion of NK cells was not associated with these clinical parameters.
It has been shown that NK cells are able to detect and eliminate tumor cells that have either lost MHC class I expression27 or upregulated stress-induced ligands.28, 29 Previously it has been suggested that the tumor cell elimination by NK cells is dysfunctional in CML not only at the diagnosis, but also during imatinib treatment.30 According to our analysis, in imatinib treated CML patients who had achieved optimal treatment response, the phenotype of NK cells was normal thus suggesting that NK cells are fully functional. This also included the expression of activating receptors (NKp30, NKp46 and NKG2D), which are known to be important in the detection of stress ligand expression on the target cells and in elimination of tumor cells. Interestingly, however, subtle phenotypic differences were observed between the groups as non-relapsing and late relapsing patients had more mature (CD57+) and cytotoxic phenotype (CD16 and CD57 expression) when compared with early-relapsing patients, and also the high proportion of CD56bright NK cells was associated with the decreased molecular relapse-free survival. The importance of NK cells in CML biology has also been demonstrated previously in studies where patients have been treated with IFN-α monotherapy.31, 32 Similarly in a study by Imagawa et al.11 it was shown that second line dasatinib-treated CML patients who successfully discontinued TKI treatment had an expansion of NK cells.
The functional studies performed confirmed that also the degranulation responses of NK cells were normal in all groups. Interestingly, the increased TNF-α/IFN-γ cytokine secretion by CD56dimCD16- NK cells upon stimulation with K562 CML cells was associated with the superior molecular relapse-free survival. It has been shown that NK cells downregulate CD16 after activation33 and in accordance a greater downregulation was observed in non-relapsing patients. Therefore, this CD56dimCD16– NK-cell subset in the non-relapsing group most likely represented CD16+ NK cells reacting to K562 cells. As such, this read-out may reflect stronger activating signaling in the groups downregulating CD16 expression. The mechanisms of how mature NK cells act against leukemia cells can either be direct, secondary, or combination of these two. It has been shown that NK-cell derived IFN-γ directs the Th1 differentiation of CD4+ T cells.34 Therefore, it was interesting to observe that in the non-relapsing patients the relative number of NK cells correlated positively with the Th1 type of cytokine secretion of CD4+ T cells.34 However, in the relapsing patients (both early and late relapsing) this correlation was negative. T- and NK-cell crosstalk has also been shown to be important for the induction of specific T-cell attack against MHC class I sufficient cell lines.35, 36
Recent advancements in the NK-cell biology have concerned the discovery of adaptive-like NK cells.37, 38 Previous studies have shown that NK cells can persist a long time and respond strongly to the same stimulus when exposed the second time.37, 38, 39, 40 Adaptive NK-cell expansions have been described in the conjunction with viral infections. In human, such adaptive NK cells lack expression of certain B-cell and myeloid-cell-related signaling proteins such as SYK, EAT-2 and FcɛRγ, in addition to the transcription factor PLZF.20, 21 Interestingly, our analyses showed that the patients who were able to stop the imatinib treatment had higher frequencies of peripheral blood EAT-2 downregulated adaptive-like NK cells. Even though no leukemia-specific adaptive NK-cell markers have yet been identified, it could be speculated that these adaptive-like NK cells defined by the markers retrieved from the viral infection studies have a role in remission, as NK cells employ a variety of activating receptors to recognize both virally infected and malignant cells. To confirm this, it would be of importance to study the response of adaptive NK cells against autologous leukemia cells in the future.
On the basis of our data, in immunological parameters late relapsing patients resemble more non-relapsing than early-relapsing group. However, a few subtle differences were noted. The expression of activating NKp30 receptor seemed to be lower in late-relapsing patients although larger numbers of patients are needed to confirm this finding. Further, the correlation between the proportion of NK cells and Th1 type CD4+ T-cell cytokine secretion was negative. This leaves room to hypothesize whether the NK cells in the late relapsing group are able to keep the disease under control for a while, but loose the battle when the situation requires the induction of adaptive T-cell responses. The division of patients into three different groups would fit well to the immunoediting theory.41 On the basis of our findings it is warranted to hypothesize that non-relapsing patients are either in the elimination or equilibrium phase where the immune system is able to keep the remaining leukemic cells under the control. In the late-relapsing group, the equilibrium state seem to hold for a while, but gradually the leukemic cells escape the control perhaps through tumor immunoediting. In the early relapsing group no specific immune response against leukemia exists at all or cells are dysfunctional and leukemia cells have escaped underneath the control of the immune system. To fully understand how residual leukemia cells modulate immune responses we would need to study their phenotype in detail. However, in the stopping scenario, the number of leukemia cells is very low or almost non-existent making the analysis of the leukemic cells very difficult.
Taken together, the functional state of the immune system is associated with the molecular relapse-free survival in CML patients discontinuing imatinib therapy. Non-relapsing patients have increased amount of NK cells, including cells with an adaptive phenotype, and their cytokine secretion is intact. Together with Th1 type T-cell responses NK cells may control the residual leukemia cells. As NK modulating agents, such as antibodies blocking inhibitory KIR, have already entered in the early clinical trials in other hematological malignancies,42 their testing in CML is warranted for increasing the proportion of patients who can discontinue imatinib treatment.
References
Hochhaus A, O'Brien SG, Guilhot F, Druker BJ, Branford S, Foroni L et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia 2009; 23: 1054–1061.
Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med 2010; 362: 2260–2270.
Saglio G, Kim DW, Issaragrisil S, le Coutre P, Etienne G, Lobo C et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362: 2251–2259.
Hoffmann VS, Baccarani M, Hasford J, Castagnetti F, Di Raimondo F, Casado LF et al. Treatment and outcome of 2904 CML patients from the EUTOS population-based registry. Leukemia 2016; e-pub ahead of print 23 September 2016; doi:10.1038/leu.2016.246.
Bhatia R, Holtz M, Niu N, Gray R, Snyder DS, Sawyers CL et al. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003; 101: 4701–4707.
Steegmann JL, Baccarani M, Breccia M, Casado LF, Garcia-Gutierrez V, Hochhaus A et al. European LeukemiaNet recommendations for the management and avoidance of adverse events of treatment in chronic myeloid leukaemia. Leukemia 2016; 30: 1648–1671.
Bjorkholm M, Ohm L, Eloranta S, Derolf A, Hultcrantz M, Sjoberg J et al. Success story of targeted therapy in chronic myeloid leukemia: a population-based study of patients diagnosed in Sweden from 1973 to 2008. J Clin Oncol 2011; 29: 2514–2520.
Mahon FX, Rea D, Guilhot J, Guilhot F, Huguet F, Nicolini F et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11: 1029–1035.
Ross DM, Branford S, Seymour JF, Schwarer AP, Arthur C, Yeung DT et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood 2013; 122: 515–522.
Etienne G, Guilhot J, Rea D, Rigal-Huguet F, Nicolini F, Charbonnier A et al. Long-term follow-up of the french stop imatinib (STIM1) study in patients with chronic myeloid leukemia. J Clin Oncol 2016; e-pub ahead of print 31 October 2016; doi:10.1200/JCO.2016.68.2914.
Imagawa J, Tanaka H, Okada M, Nakamae H, Hino M, Murai K et al. Discontinuation of dasatinib in patients with chronic myeloid leukaemia who have maintained deep molecular response for longer than 1 year (DADI trial): a multicentre phase 2 trial. Lancet Haematol 2015; 2: e528–e535.
Youden WJ . Index for rating diagnostic tests. Cancer 1950; 3: 32–35.
Salih J, Hilpert J, Placke T, Grunebach F, Steinle A, Salih HR et al. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int J Cancer 2010; 127: 2119–2128.
Schade AE, Schieven GL, Townsend R, Jankowska AM, Susulic V, Zhang R et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood 2008; 111: 1366–1377.
Seggewiss R, Lore K, Greiner E, Magnusson MK, Price DA, Douek DC et al. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood 2005; 105: 2473–2479.
Rousselot P, Huguet F, Rea D, Legros L, Cayuela JM, Maarek O et al. Imatinib mesylate discontinuation in patients with chronic myelogenous leukemia in complete molecular remission for more than 2 years. Blood 2007; 109: 58–60.
Farag SS, Caligiuri MA . Human natural killer cell development and biology. Blood Rev 2006; 20: 123–137.
Kared H, Martelli S, Ng TP, Pender SL, Larbi A . CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother 2016; 65: 441–452.
Cooper MA, Fehniger TA, Caligiuri MA . The biology of human natural killer-cell subsets. Trends Immunol 2001; 22: 633–640.
Schlums H, Cichocki F, Tesi B, Theorell J, Beziat V, Holmes TD et al. Cytomegalovirus infection drives adaptive epigenetic diversification of NK cells with altered signaling and effector function. Immunity 2015; 42: 443–456.
Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015; 42: 431–442.
Grzywacz B, Kataria N, Verneris MR . CD56(dim)CD16(+) NK cells downregulate CD16 following target cell induced activation of matrix metalloproteinases. Leukemia 2007; 21: 356–359.
Zheng LM, Ojcius DM, Garaud F, Roth C, Maxwell E, Li Z et al. Interleukin-10 inhibits tumor metastasis through an NK cell-dependent mechanism. J Exp Med 1996; 184: 579–584.
Baccarani M, Efficace F, Rosti G . Moving towards patient-centered decision-making in chronic myeloid leukemia: assessment of quality of life and symptom burden. Haematologica 2014; 99: 205–208.
Hughes TP, Ross DM . Moving treatment-free remission into mainstream clinical practice in CML. Blood 2016; 128: 17–23.
Saussele S, Richter J, Hochhaus A, Mahon FX . The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016; 30: 1638–1647.
Karre K, Ljunggren HG, Piontek G, Kiessling R . Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986; 319: 675–678.
Diefenbach A, Jensen ER, Jamieson AM, Raulet DH . Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 2001; 413: 165–171.
Cerwenka A, Baron JL, Lanier LL . Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc Natl Acad Sci USA 2001; 98: 11521–11526.
Chen CI, Koschmieder S, Kerstiens L, Schemionek M, Altvater B, Pscherer S et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR-ABL-positive mice. Leukemia 2012; 26: 465–474.
Ilander M, Kreutzman A, Rohon P, Melo T, Faber E, Porkka K et al. Enlarged memory T-cell pool and enhanced Th1-type responses in chronic myeloid leukemia patients who have successfully discontinued IFN-alpha monotherapy. PLoS ONE 2014; 9: e87794.
Kreutzman A, Rohon P, Faber E, Indrak K, Juvonen V, Kairisto V et al. Chronic myeloid leukemia patients in prolonged remission following interferon-alpha monotherapy have distinct cytokine and oligoclonal lymphocyte profile. PLoS ONE 2011; 6: e23022.
Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood 2013; 121: 3599–3608.
Martin-Fontecha A, Thomsen LL, Brett S, Gerard C, Lipp M, Lanzavecchia A et al. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat Immunol 2004; 5: 1260–1265.
Kelly JM, Darcy PK, Markby JL, Godfrey DI, Takeda K, Yagita H et al. Induction of tumor-specific T cell memory by NK cell-mediated tumor rejection. Nat Immunol 2002; 3: 83–90.
Kelly JM, Takeda K, Darcy PK, Yagita H, Smyth MJ . A role for IFN-gamma in primary and secondary immunity generated by NK cell-sensitive tumor-expressing CD80 in vivo. J Immunol 2002; 168: 4472–4479.
Foley B, Cooley S, Verneris MR, Pitt M, Curtsinger J, Luo X et al. Cytomegalovirus reactivation after allogeneic transplantation promotes a lasting increase in educated NKG2C+ natural killer cells with potent function. Blood 2012; 119: 2665–2674.
Sun JC, Beilke JN, Lanier LL . Adaptive immune features of natural killer cells. Nature 2009; 457: 557–561.
Sun JC, Beilke JN, Bezman NA, Lanier LL . Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med 2011; 208: 357–368.
Lopez-Verges S, Milush JM, Schwartz BS, Pando MJ, Jarjoura J, York VA et al. Expansion of a unique CD57(+)NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA 2011; 108: 14725–14732.
Dunn GP, Old LJ, Schreiber RD . The three Es of cancer immunoediting. Annu Rev Immunol 2004; 22: 329–360.
Vey N, Bourhis JH, Boissel N, Bordessoule D, Prebet T, Charbonnier A et al. A phase 1 trial of the anti-inhibitory KIR mAb IPH2101 for AML in complete remission. Blood 2012; 120: 4317–4323.
Acknowledgements
We thank patients, study nurses and other personnel in the clinical centers for their participation in this trial. This work was supported by the Nordic Cancer Union, Finnish Society of Hematology, Biomedicum Helsinki Foundation, Research Foundation of Blood Diseases in Finland, Academy of Finland, Finnish Cancer Organizations, Signe and Ane Gyllenberg Foundation, Finnish Cancer Institute and Doctoral Programme in Clinical Research in the University of Helsinki.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
UOS has received honoraria from Ariad. PK has received honoraria from Novartis, BMS, Ariad and Pfizer. HE has received development grant from Novartis and travel grants form BMS and Novartis. HHjH has received honoraria from Novartis, Bristol-Myers Squibb and Ariad. JR has received honoraria and research funding from Novartis and Bristol-Myers Squibb and honoraria from Ariad. SM and KP have received honoraria and research funding from Novartis, Bristol-Myers Squibb, and Pfizer. SM has received research funding from Ariad. The remaning authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ilander, M., Olsson-Strömberg, U., Schlums, H. et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 31, 1108–1116 (2017). https://doi.org/10.1038/leu.2016.360
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2016.360
