
Abstract
In this open-label, intra-patient phase I/II trial, bortezomib was replaced with carfilzomib (escalated from 20 to 45 mg/m2 on days 1, 2, 8, 9, 15 and 16 of a 28-day cycle) for multiple myeloma (MM) patients who progressed while on or within 12 weeks of receiving a bortezomib-containing combination regimen. Study objectives included determination of the maximum tolerated dose (MTD), overall response rate (ORR), clinical benefit rate (CBR), time to progression, time to response, duration of response, progression-free survival and overall survival (OS). Of 38 registered patients, 37 were treated and evaluable for efficacy and safety. Thirty-one carfilzomib-based regimens using 14 different drug combinations were tested. One regimen (carfilzomib (45 mg/m2), ascorbic acid (1000 mg) and cyclophosphamide (2.2 mg/kg)) reached MTD. ORR and CBR were 43.2 and 62.2%, respectively. Median progression-free survival, time to progression and OS were 8.3, 9.9 and 15.8 months, respectively. Hematologic adverse events (AEs; ⩾grade 3) included lymphopenia (35.1%), thrombocytopenia (24.3%), anemia (10.8%) and neutropenia (10.8%). Nonhematologic AEs (⩾grade 3) included fever (5.4%) and hypokalemia (5.4%). These results demonstrate that replacing bortezomib with carfilzomib is safe and can be effective for MM patients failing bortezomib-containing combination regimens. This trial was registered at http://www.clinicaltrials.gov (#NCT01365559).
Similar content being viewed by others


Introduction
In 2013, ∼22 500 individuals in the United States were diagnosed with multiple myeloma (MM), and nearly 11 000 died from the disease.1 Although the use of novel agents and high-dose therapy has improved overall survival (OS),2 MM remains incurable; nearly all patients relapse or become refractory to treatment. Thus, more treatment combinations are needed, especially for those patients who have exhausted all available treatment options.3
In July 2012, the proteasome inhibitor (PI) carfilzomib was approved as a single agent in the United States for the treatment of relapsed and refractory MM based on the findings from the pivotal PX-171-003-A1 (#NCT00511238)) trial.4, 5 In this study, nearly all patients who received carfilzomib had received prior therapy with bortezomib, a first-generation PI.4 Among patients who were refractory or intolerant to bortezomib and lenalidomide, the ORR was 20.1% (95% confidence interval (CI): 14.9–26.1). Similarly, many relapsed and refractory MM patients who received single-agent carfilzomib in the PX-171-004 study (#NCT00530816) had also received bortezomib and had an ORR of 17.1%.6 These results suggest the potential for this newer PI to overcome resistance to other PIs such as bortezomib. This type of strategy has proven to be successful, as MM patients failing regimens that include steroids and an immunomodulatory drug (IMiD) often respond to a different IMiD.7
The success achieved following retreatment of patients with bortezomib has occurred in specific clinical settings. Bortezomib retreatment has been successful for some patients that were previously responsive to this PI,8, 9, 10 and for relapsed and refractory MM patients treated with it in combination with other anti-MM agents.11, 12 The ability to overcome bortezomib resistance following retreatment with a different PI has been demonstrated in preclinical studies with bortezomib, carfilzomib, oprozomib (ONX 0912), ixazomib (MLN 9708), delanzomib (CEP-18770) and MLN 2238. For instance, MLN 2238 was shown to induce apoptosis in bortezomib-resistant MM cells.13 Carfilzomib in combination with marizomib (NPI-0052) was also found to induce cell death in MM cells that were derived from bortezomib refractory patients.14, 15 In a separate study, the PI delanzomib had anti-MM effects in bortezomib-resistant MM, and the combination of delanzomib and bortezomib was found to have greater anti-MM activity compared with either agent alone.16 These preclinical studies provided the rationale for evaluating whether replacing a PI for one that is ineffective in a combination treatment can achieve clinical efficacy for patients with MM. In this context, carfilzomib treatment for patients that failed bortezomib therapy in an otherwise novel treatment combination has shown clinical activity,4, 6 yet the concept of substituting PI with another into an otherwise unchanged combination treatment in an attempt to overcome PI resistance has not been investigated in the clinic. Given that single-agent carfilzomib can produce responses in some bortezomib refractory patients4 and shows high response rates when combined with other agents,17, 18 we hypothesized that carfilzomib would be an effective and well-tolerated replacement for bortezomib among MM patients who had failed bortezomib-containing combination regimens. In this study, we investigated the feasibility of replacing bortezomib with carfilzomib in an otherwise identical combination regimen for MM patients who had progressed on or within 12 weeks of receiving their most recent bortezomib-based combination regimen.
Patients and methods
This was a multicenter, open-label, nonrandomized, intra-patient dose-escalating phase I/II clinical study that involved seven sites in the United States. This study was conducted in accordance with US Food and Drug Administration regulations, the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. The study was compliant with all local health authority and Institutional Review Board requirements. All patients provided written informed consent in accordance with federal, local and institutional guidelines. The investigators designed the study and collected and analyzed data from the study. All participating institutions received financial assistance from Onyx Pharmaceuticals Inc. (South San Francisco, CA, USA).
Patients
MM patients ⩾18 years old with measurable and progressive disease and who had progressed while on or within 12 weeks of receiving the last dose of their most recent bortezomib-containing regimen were eligible. All patients must have received four doses of bortezomib (⩾1.0 mg/m2) in 21 or 28 days per cycle for at least one cycle. In addition to bortezomib, a patient’s most recent bortezomib-containing regimen must have contained an alkylating agent (for example, melphalan, cyclophosphamide or bendamustine), anthracycline (for example, doxorubicin or pegylated liposomal doxorubicin), IMiD (thalidomide and/or lenalidomide) and/or glucocorticosteroid (prednisone, dexamethasone or methylprednisolone). Patients were required to have not received any anti-MM therapy for ⩾3 weeks before enrollment to ensure that there was adequate washout of the prior anti-myeloma regimen (see below).
Patients must have had a life expectancy of ⩾3 months and an Eastern Cooperative Oncology Group performance status of 0–2. Laboratory entry criteria included an absolute neutrophil count of ⩾1.5 × 109/l (⩾1.0 × 109/l if the bone marrow was extensively infiltrated); hemoglobin count of ⩾8 g/dl; platelet count of ⩾75 × 109/l (⩾50 × 109/l if the bone marrow was extensively infiltrated); creatinine clearance ⩾30 ml/min; aspartate transaminase (serum glutamic oxaloacetic transaminase) and ALT (serum glutamic-pyruvic transaminase) ⩽3 × upper limit of normal or ⩽5 × upper limit of normal if hepatic metastases were present; serum total bilirubin ⩽1.5 × upper limit of normal and serum potassium ⩾3 and ⩽5. Men were required to use contraception; women of childbearing potential were also required to use contraception and receive pregnancy tests throughout the study.
Patients were excluded if they had peripheral neuropathy (grades 3 or 4 or grade 2 with pain) within 14 days before enrollment; impaired cardiac function or clinically significant cardiac disease (for example, unstable angina or myocardial infarction within 4 months before enrollment; New York Heart Association Class III or IV congestive heart failure (CHF); uncontrolled angina; clinically significant pericardial disease or arrhythmias); gastrointestinal disease or impaired gastrointestinal function; plasma cell dyscrasia with polyneuropathy, organomegaly, endocrinopathy, M-protein and skin changes syndrome; plasma cell leukemia; a prior malignancy within 5 years except non-melanomatous skin cancer; major surgery within 28 days before enrollment or severe hypercalcemia (corrected serum calcium ⩾12 mg/dl). Patients were also excluded if they received prior therapy with carfilzomib or chemotherapy, corticosteroids (>10 mg per day prednisone or equivalent), immunotherapy, antibody therapy, thalidomide, lenalidomide, arsenic trioxide, bortezomib or radiation therapy within 21 days of enrollment; other experimental drug or therapy within 28 days of enrollment or concurrent use of any other anticancer agents or treatments. In addition, patients were excluded if they had experienced an acute active infection that required treatment, known human immunodeficiency virus infection or active hepatitis B, or C infection. Pregnant or lactating females were also excluded.
Study design and drug administration
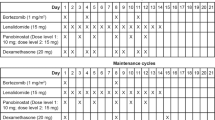
The study design is shown in Figure 1. The primary objectives of the study were to determine the maximum tolerated dose (MTD) for each combination regimen and the ORR for all regimens. The regimen, dose and schedule of the prior bortezomib-containing regimen remained unchanged except that bortezomib was replaced with carfilzomib. For three patients, cycle length was extended from 21 to 28 days by adding a week without additional treatment in order to conform to the carfilzomib schedule. During cycle 1, a starting dose of carfilzomib (20 mg/m2) was given by intravenous administration over 30 min on days 1, 2, 8, 9, 15 and 16 of a 28-day cycle. During cycles 2, 3 and 4, carfilzomib was escalated to 27, 36 and 45 mg/m2, respectively; the same infusion time and dosing schedule that was used during cycle 1 was also used during cycles 2–4.

Trial design and dosing schema. (a) The trial replicates patients’ last bortezomib-containing combination regimen except that bortezomib is replaced with carfilzomib. The other agent(s), dose and administration schedule in the regimen remain the same as the prior bortezomib-containing regimen. (b) Carfilzomib dosing and schedule for the study treatment and maintenance periods. aAlkylating agents with or without steroids; glucocorticoids; IMiDs with or without other agents; and PLD with or without steroids. bPatients underwent intra-patient dose escalation during cycles 1–4, unless a DLT occurred. Following the protocol amendment, if a DLT occurred, the patient continued dosing at the dose administered in the previous cycle. cThe carfilzomib dose in cycles 5–8 was the highest tolerated dose received in cycles 1–4 without DLTs, or significant AEs. dFor maintenance, carfilzomib was administered as a single agent at the last dose that the patient was receiving when he or she completed the combination study treatment. AE, adverse event; DLT, dose-limiting toxicity; IMiD, immunomodulatory drug; PLD, pegylated liposomal doxorubicin.
During cycle 1, all patients received oral or intravenous fluids (250–500 ml) before each dose of carfilzomib. Patients also received prophylactic acyclovir (or another similar anti-varicella agent). Bisphosphonates and erythropoietic agents were permitted during the study. Colony-stimulating factors were permitted if grade 4 neutropenia occurred but could not be used prophylactically. During cycle 1, oral or intravenous dexamethasone (4 mg) was given before each carfilzomib dose unless the patient was already receiving glucocorticosteroids on the same day as a part of their anti-MM treatment regimen. Patients who were believed to be at risk for tumor lysis syndrome (TLS) were allowed to receive allopurinol prophylaxis.
During cycles 1–4, patients were required to complete a full cycle without experiencing a dose-limiting toxicity (DLT) before they could begin a new cycle. During cycles 5–8, the maximum dose that the patient tolerated during the first four cycles was given using the same dosing and administration schedule. Each of the first 19 patients continued to receive treatment until one of the following occurred: a DLT, a maximum response plus one additional treatment cycle was completed, a maximum of eight treatment cycles was received, disease progression (PD), or unacceptable toxicity. After enrolling the first 19 patients, the study protocol was amended to allow the following: (1) up to two carfilzomib dose reductions (9 mg/m2 per dose reduction) for patients that experienced a DLT (previous to approval of this amendment, patients experiencing DLTs were taken off the trial); and (2) maintenance therapy with single-agent carfilzomib for patients that completed eight cycles of treatment without PD. For maintenance treatment, carfilzomib was administered as a single agent on days 1, 2, 15 and 16 of each 28-day cycle at the last dose that the patient was receiving when he or she completed the combination study treatment. Once a patient stopped receiving study drug (carfilzomib), follow-up for PD and OS was completed every 3 and 6 months, respectively.
Dose-limiting toxicities
DLTs were defined as the occurrence of any of the following during the first four cycles of dose escalation treatment: grade 3 or 4 hematologic toxicity (excluding lymphopenia); grade 3 thrombocytopenia with grade 3 or 4 hemorrhage; grade 3 or 4 nausea or vomiting refractory to anti-emetic therapy; grade 3 or 4 diarrhea or constipation refractory to antidiarrheal or constipation therapy; any other study treatment-related⩾grade 2 nonhematologic toxicity (except alopecia); or any treatment-related death.
Determination of the MTD
MTD declaration required a minimum of three patients for each specific combination treatment. If all three patients showed no DLTs during the first four cycles of dose escalation to 45 mg/m2, the MTD was declared at this maximally administered dose (MAD). If a DLT occurred in one subject at any dose level during any of the first four dose escalation cycles, additional subjects were enrolled at this dose level until either another DLT was observed or a total of six patients were treated without an additional DLT. If no additional DLTs occurred among six patients treated, then the MTD was considered to be the MAD. However, if more than one patient experienced a DLT, then the MTD was declared at one dose level lower than that at which the second DLT occurred as long as three patients had been treated at the lower level without a DLT or no more than one DLT occurred among six patients.
Safety assessments
Adverse events (AEs) were assessed for any patient receiving study drug at each visit and graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0. Serious AEs included the following: any death occurring within 28 days of receiving study drug regardless of discontinuation from the study; a life-threatening experience that the investigator believed placed the participant at immediate risk of death; an event requiring inpatient hospitalization or prolongation of an existing hospitalization (excluding scheduled hospitalizations for nonacute medical conditions or elective surgery) or a condition that resulted in persistent or significant disability/incapacity.
Response criteria
Response and PD were determined according to a modified version of the European Blood and Marrow Transplantation criteria for evaluating disease response and progression among MM patients and the updated International Myeloma Working Group criteria.19, 20 The ORR (complete response+very good partial response+partial response) and clinical benefit rate (CBR; ⩾minimal response (MR)) were determined. Disease assessment occurred during the last week of the cycle. If a patient experienced ⩾MR, assessment of disease response was performed 4 weeks later to confirm the patient’s response to treatment.
Statistical methods
The intent-to-treat population included all patients who registered and met eligibility criteria. The safety and efficacy population included all patients who received ⩾1 dose of study drug. ORR estimates were accompanied by a two-sided 95% exact CI that used a binomial distribution. PD had to be confirmed in all cases but was recorded when laboratory tests first showed PD. Patients who did not meet protocol-defined PD but were taken off study owing to worsening disease were treated as if they were taken off study for PD. Median time to progression, duration of response, progression-free survival and OS were estimated using Kaplan–Meier methods. Median follow-up time was estimated using the reverse Kaplan–Meier method.21 P-values for patient response were calculated using Fisher’s test (two tails). Statistical analyses were performed using SPSS version 21 (IBM, Armonk, NY, USA) or GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA).
Results
Patient disposition
From 2 May 2011 to 14 January 2013, 38 patients entered the intent-to-treat population, but 1 patient chose not to receive study treatment following registration. Thus, the efficacy and safety populations both consisted of 37 patients. The cutoff date for this data analysis was 25 April 2013. At data cutoff, 13 patients were still active on the trial, 5 patients were receiving active combination treatment and 8 were receiving maintenance with single-agent carfilzomib.
Baseline demographics and clinical characteristics are summarized in Table 1. Median time from diagnosis of MM to study enrollment was 4.2 years (range, 5 months–10.5 years), and patients had received a median of five (range, 1–23) prior lines of anti-MM therapy. Patients had received a median of two (range, 1-13) different prior bortezomib-containing regimens. Dosage and schedule of the last bortezomib-containing regimens are outlined in Supplementary Table S1. Only 26.3 and 15.8% of patients had achieved ⩾MR or ⩾PR, respectively, on the bortezomib-containing regimen that they eventually failed. Patients had IgG (55.3%), IgA (21.1%) or light-chain-only (21.1%) MM. At baseline, 15.8% of patients had peripheral neuropathy (any grade).
Treatment exposure and treatment discontinuations
Patients received 31 unique treatment regimens throughout the trial (Table 1 and Supplementary Table S1). One patient who withdrew consent before receiving study treatment had previously been treated with bortezomib and lenalidomide. The 31 specific treatment regimens stemmed from 14 different drug combinations across all four active anti-MM drug classes (Table 2 and Supplementary Table S2). Drug combinations included carfilzomib and the following agents: dexamethasone alone (31.6% of patients); the alkylating agents melphalan, cyclophosphamide or bendamustine (28.9%) with or without steroids; pegylated liposomal doxorubicin (18.4%) with or without steroids; and/or the IMiDs thalidomide and/or lenalidomide (21.1%) with or without other agents. More than half of patients (59.5%) received the highest planned carfilzomib dose (45 mg/m2; Supplementary Table S2). The median duration of carfilzomib treatment was four cycles (range, <1–8 cycles). Eighteen patients completed eight cycles of treatment and chose to continue receiving single-agent carfilzomib maintenance therapy.
Twenty-five patients discontinued study treatment for the following reasons: nine because of PD; one because of death; one was removed because of the worsening of MM-related symptoms thought to be related to PD; five owing to withdrawal of informed consent because of personal reasons (n=3) or AEs that interfered with quality of life (n=2); seven owing to DLTs (thrombocytopenia (n=2); abdominal pain (n=1); CHF (n=1); TLS (n=1); neutropenia (n=1); and febrile neutropenia (n=1)); and two owing to AEs (intercurrent illness (n=1) and sepsis (n=1)). Twenty-five patients progressed, including 10 patients who progressed after showing an initial response.
Safety
DLTs and determination of the MTD
Eight carfilzomib-based drug combinations led to the occurrence of 10 DLTs across a variety of combination treatments (Supplementary Table S2), including grade 4 thrombocytopenia (n=4); grade 3 neutropenia (n=2); grade 3 sepsis (n=1); grade 3 CHF (n=1); grade 3 TLS (n=1); and grade 2 abdominal pain (n=1). Seven patients were taken off the study owing to DLTs and three were removed before the study protocol amendment allowing carfilzomib dose reduction was approved (Supplementary Table S2). After the amendment, four patients were removed after completion of one (n=2), two (n=1) or three cycles (n=1) of treatment. Of the 31 treatment regimens tested, the MTD was reached at MAD for the regimen that contained carfilzomib (45 mg/m2), ascorbic acid (1000 mg) and cyclophosphamide (2.2 mg/kg; Supplementary Table S2).
Treatment-emergent AEs
The most common hematologic and nonhematologic treatment-emergent AEs across all patients are shown in Table 3. Hematologic AEs⩾grade 3 included lymphopenia (35.1%), thrombocytopenia (24.3%), anemia (10.8%), neutropenia (10.8%), leukopenia (5.4%) and decreased red blood cell count (2.7%). Lymphopenia (⩾grade 3) occurred in all five patients who received bendamustine. Overall, five patients (13.5%) experienced grade 4 hematologic AEs, including thrombocytopenia (10.8%) and lymphopenia (5.4%). One patient experienced both of these grade 4 AEs. Only one patient experienced a hematologic serious AE (febrile neutropenia).
Nonhematologic AEs ⩾grade 3 included fever (5.4%), hypokalemia (5.4%), increased creatinine (2.7%), chills (2.7%), hypertension (2.7%), migraine (2.7%), dehydration (2.7%), pneumonia (2.7%), urinary tract infection (2.7%), hypoechoic liver lesions (2.7%), sepsis (2.7%), steel plate owing to osteonecrosis of the jaw (2.7%), TLS (2.7%) and hyponatremia (2.7%). Peripheral neuropathy was infrequent (16%) and all cases were ⩽grade 2 (grade 1 and grade 2 (n=3 each)).
There were 12 (32.4%) nonhematologic serious AEs, including the following: rectal bleeding (grade 2); cellulitis, dehydration, fever with chills, generalized weakness, influenza, pruritus, renal dysfunction/failure, TLS and urinary tract infection with fever (all grade 3); tachyarrhythmia (grade 4); and pneumonia (grade 5). There was one grade 4 nonhematologic AE (sepsis).
Of the 21 patients that were not removed from the trial because of DLTs, AEs or informed consent withdrawal, treatment was held in three patients (14.3%), two (9.5%) of whom experienced a subsequent dose reduction.
Efficacy
Among the 37 treated patients, the ORR was 43.2% and the CBR was 62.2% with responses observed across all drug classes (Table 4). Specifically, among patients (n=12) who received carfilzomib with dexamethasone alone, the ORR was 33.3% and the CBR was 58.3%. The ORR was 54.5% and the CBR was 63.6% for patients (n=11) who were given carfilzomib and an alkylating agent with or without steroids. For those patients who received carfilzomib and an IMiD with or without other agents (n=7), the ORR was 42.9% and the CBR was 57.1%. For patients (n=7) who received carfilzomib and pegylated liposomal doxorubicin with or without steroids, the ORR was 42.9% and the CBR was 71.4%. Five patients (13.5%) who were not evaluable for response (two patients had no confirmed response; three patients suffered DLTs in cycle 1 and were either hospitalized or never completed a full cycle) were considered as not responsive to treatment.
There was a trend, albeit not significant for patients who had achieved ⩾MR on the bortezomib-containing version of the regimen (before developing PD) to respond better to treatment with the carfilzomib-containing version (⩾PR, 60.0%) than patients who did not (⩾PR, 36.8%; P=0.5097). The proportion of patients achieving ⩾MR with the carfilzomib-containing regimen who had previously been treated with 1.0 mg/m2 bortezomib (63.6%) was similar to that of patients with ⩾MR who had received ⩾1.3 mg/m2 bortezomib (53.3%; P=0.7893).
Progression-free survival, OS and duration of response are shown in Figure 2. Median follow-up time was 13.7 months (range, 1.2–19.4; Table 4). Median progression-free survival was 8.3 months (95% CI: 4.6–12.1), and median OS was 15.8 months (95% CI: 13.5–18.3). Median time to progression was 9.9 months (95% CI: 6.7–13.1). Median duration of response among patients who achieved ⩾PR (43.2%) and⩾MR (62.2%) was 9.9 months (95% CI: 7.6–12.2) and 9.2 months (95% CI: 6.7–11.7), respectively. A summary of time-to-event outcomes is shown in Table 5. Median time to first response (⩾MR) was 1.6 months (95% CI: 0.55–2.68), and the median time to best response was 3.4 months (95% CI: 2.7–4.1).

The Kaplan–Meier curve for time-to-event data for all evaluable patients. (a) PFS in months. (b) OS in months. (c) DOR in months. DOR, duration of response; OS, overall survival; PFS, progression-free survival.
Discussion
Our study demonstrates that carfilzomib can be used safely and effectively in place of bortezomib when combined with many other classes of drugs including alkylators, anthracyclines, IMiDs and glucocorticosteroids. In addition, this trial shows that carfilzomib is often an effective replacement for bortezomib for patients with MM who progressed from the same combination treatment containing bortezomib. Concerns about safety and tolerability and drug–drug interactions often arise whenever new agents are introduced into combination regimens, particularly in heavily pretreated patients who are more likely to be unfit or frail compared with patients with newly diagnosed MM. The breadth of different regimens (n=31) that were evaluated in our study suggests that carfilzomib can be combined with the other classes of drugs that have shown efficacy for treating MM.
In our study, the AE profile of the carfilzomib-containing regimens was similar to that observed in 526 patients who were previously enrolled in four phase II clinical studies (PX-171-003-A0, 003-A1, PX-171-004 and PX-171-005) and received single-agent carfilzomib.22 In our study, lymphopenia, thrombocytopenia, anemia and neutropenia were the most common ⩾grade 3 hematologic AEs, which is similar to those observed with single-agent carfilzomib.22 Fever and hypokalemia were the most common ⩾grade 3 nonhematologic AEs in our study, whereas pneumonia, fatigue and dyspnea were the most common ⩾grade 3 nonhematologic AEs with single-agent carfilzomib.22 One patient (2.7%) had CHF, which compares favorably with the four phase II studies that investigated single-agent carfilzomib, in which 30 patients (5.7%) had ⩾grade 3 CHF. Dyspnea was not observed in our study.
The trial involved intra-patient dose escalation of carfilzomib (20, 27, 36 and 45 mg/m2) during the first four cycles of treatment. More than half (59.5%) of the treated patients received the MAD of carfilzomib (45 mg/m2). Because of the large number (n=31) of different carfilzomib-containing combination treatments among the 37 enrolled patients, the MTD was only established for one regimen (ascorbic acid (1000 mg) and cyclophosphamide (2.2 mg/kg)) at the MAD of carfilzomib.
Overall, 14 different combinations of drugs using agents from four therapeutic classes were tested. At least one patient in 11 (78.8%) of these 14 drug combinations achieved ⩾MR. Similar CBRs were observed among the four therapeutic classes combined with carfilzomib. The observation of clinical benefit across all effective anti-MM agent classes is promising and is consistent with recent studies showing that carfilzomib is safe and effective when used in combination with a variety of drugs, including lenalidomide and dexamethasone,17, 18, 23, 24, 25 pomalidomide and dexamethasone,26 melphalan and prednisone,27 cyclophosphamide and dexamethasone,28 thalidomide and dexamethasone,29 and cyclophosphamide, thalidomide and dexamethasone.30 Results from those studies also compare favorably with bortezomib-based regimens, including bortezomib in combination with lenalidomide and dexamethasone.31, 32 Although the latter combination yielded a high response rate, 64% of patients developed grade 1 or grade 2 peripheral neuropathy, and 62% of patients required a dose reduction.32 A recent clinical trial evaluating carfilzomib administered at 45 and 56 mg/m2 in combination with dexamethasone showed an ORR of 55% and good tolerability for relapsed and refractory MM patients without bortezomib resistance.25 Our study extends these findings and demonstrates that carfilzomib at doses up to 45 mg/m2 and combined with dexamethasone is efficacious and has an acceptable safety profile for MM patients who are refractory to bortezomib.
The mechanisms by which carfilzomib produces responses for patients who have failed bortezomib-based combination treatments is unclear, but it may partially depend on differences in the chemical properties and mode of action of these two PIs. For example, bortezomib is a reversible PI, whereas carfilzomib binds selectively and irreversibly to the proteasome.14, 33, 34 In addition, there may be differences in the synergistic effects between bortezomib and other agents relative to carfilzomib. The specific mechanisms that cause bortezomib resistance35 will need to be more fully elucidated to determine the underlying basis for carfilzomib’s ability to overcome bortezomib resistance.
Although it has been clearly shown that patients resistant to a bortezomib-containing combination regimen often respond to other agents combined with bortezomib, this is the first trial to demonstrate that a different PI (carfilzomib) can overcome resistance to bortezomib when replacing it in the same combination treatment. Findings from our trial suggest that carfilzomib can be safely and effectively used as a replacement for bortezomib across a wide variety of anti-MM regimens to treat patients that have failed prior bortezomib-containing regimens. However, this study was not designed to determine which specific carfilzomib-containing treatment combinations have better safety or efficacy profiles in the setting of bortezomib resistance, and further phase 1 and 2 studies testing the most promising combinations will be required.
Although we succeeded in establishing that replacement of bortezomib with carfilzomib can be effective across many different combination regimens, we only determined the MTD for one regimen. Nevertheless, more than half of all treated patients received the MAD for carfilzomib (45 mg/m2). The dose and schedule of bortezomib used in the different combination treatments that patients had failed varied, which could have potentially affected the efficacy of subsequent treatment with carfilzomib. However, our data did not reveal any bias toward responses being more frequently observed with a specific prior bortezomib dose or schedule. Along with others, our group has shown that different doses (1 or 1.3 mg/m2) and schedules (3 or 4 weeks per cycle; Supplementary Table S1) of bortezomib produce similar results when used either as a single-agent or in combination with other agents.11, 12, 36 Moreover, although patients may initially be treated at a dose of 1.3 mg/m2, many patients require reductions in their dose to 1.0 mg/m2, weekly dosing or longer cycles because of toxicity.37 Overall, the results demonstrate that carfilzomib can safely and effectively replace bortezomib for the treatment of MM patients who progress while on or within 12 weeks of failing a wide range of bortezomib-containing combination regimens. Expanded phase 1 and 2 trials will help characterize the activity and tolerability of several of the most promising treatment combinations revealed in this study. By partnering with new agents, including those that are from the same therapeutic class as the agent that is being replaced, patients may be able to overcome treatment resistance and expand their therapeutic options. Similar results have been observed among MM patients who were previously treated with IMiDs.7 Thus, the findings from this trial suggest the same phenomenon occurs with PIs and should provide clinicians with a new therapeutic option: replacement of bortezomib with carfilzomib for patients who fail a bortezomib-containing combination regimen.
References
Cancer Facts & FiguresAm Cancer Soc 2013.
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008; 111: 2516–2520.
Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia 2012; 26: 149–157.
Siegel DS, Martin T, Wang M, Vij R, Jakubowiak AJ, Lonial S et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012; 120: 2817–2825.
Onix Pharmaceuticals, IncSouth San Francisco C. Krypolis Prescribing Information 2013.
Vij R, Siegel DS, Jagannath S, Jakubowiak AJ, Stewart AK, McDonagh K et al. An open-label, single-arm, phase 2 study of single-agent carfilzomib in patients with relapsed and/or refractory multiple myeloma who have been previously treated with bortezomib. Br J Haematol 2012; 158: 739–748.
Madan S, Lacy MQ, Dispenzieri A, Gertz MA, Buadi F, Hayman SR et al. Efficacy of retreatment with immunomodulatory drugs (IMiDs) in patients receiving IMiDs for initial therapy of newly diagnosed multiple myeloma. Blood 2011; 118: 1763–1765.
Sood R, Carloss H, Kerr R, Lopez J, Lee M, Druck M et al. Retreatment with bortezomib alone or in combination for patients with multiple myeloma following an initial response to bortezomib. Am J Hematol 2009; 84: 657–660.
Taverna C, Voegeli J, Trojan A, Olie Ra, von Rohr A . Effective response with bortezomib retreatment in relapsed multiple myeloma—a multicentre retrospective survey in Switzerland. Swiss Med Wkly 2012; 142: w13562.
Petrucci MT, Giraldo P, Corradini P, Teixeira A, Dimopoulos MA, Blau IW et al. A prospective, international phase 2 study of bortezomib retreatment in patients with relapsed multiple myeloma. Br J Haematol 2013; 160: 649–659.
Berenson JR, Yellin O, Kazamel T, Hilger JD, Chen C-S, Cartmell a et al. A phase 2 study of pegylated liposomal doxorubicin, bortezomib, dexamethasone and lenalidomide for patients with relapsed/refractory multiple myeloma. Leukemia 2012; 26: 1675–1680.
Berenson JR, Yellin O, Bessudo A, Boccia RV, Noga SJ, Gravenor DS et al. Phase I/II trial assessing bendamustine plus bortezomib combination therapy for the treatment of patients with relapsed or refractory multiple myeloma. Br J Haematol 2013; 160: 321–330.
Chauhan D, Tian Z, Zhou B, Kuhn D, Orlowski R, Raje N et al. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin Cancer Res 2011; 17: 5311–5321.
Kuhn DJ, Chen Q, Voorhees PM, Strader JS, Shenk KD, Sun CM et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood 2007; 110: 3281–3290.
Fenical W, Jensen PR, Palladino MA, Lam KS, Lloyd GK, Potts BC . Discovery and development of the anticancer agent salinosporamide A (NPI-0052). Bioorg Med Chem 2009; 17: 2175–2180.
Sanchez E, Li M, Li J, Wang C, Chen H, Jones-Bolin S et al. CEP-18770 (delanzomib) in combination with dexamethasone and lenalidomide inhibits the growth of multiple myeloma. Leuk Res 2012; 36: 1422–1427.
Jakubowiak AJ, Dytfeld D, Griffith KA, Lebovic D, Vesole DH, Jagannath S et al. A phase 1/2 study of carfilzomib in combination with lenalidomide and low-dose dexamethasone as a frontline treatment for multiple myeloma. Blood 2012; 120: 1801–1809.
Wang M, Martin T, Bensinger W, Alsina M, Siegel DS, Kavalerchik E et al. Phase 2 dose-expansion study (PX-171-006) of carfilzomib, lenalidomide and low-dose dexamethasone in relapsed or progressive multiple myeloma. Blood 2013; 122: 3122–3128.
BladÉ J, Samson D, Reece D, Apperley J, BJÖrkstrand B, Gahrton Gö et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Br J Haematol 1998; 102: 1115–1123.
Durie BGM, Harousseau J-L, Miguel JS, Bladé J, Barlogie B, Anderson K et al. International uniform response criteria for multiple myeloma. Leukemia 2006; 20: 1467–1473.
Schemper M, Smith TL . A note on quantifying follow-up in studies of failure time. Control Clin Trials 1996; 17: 343–346.
Siegel D, Martin T, Nooka A, Harvey RD, Vij R, Niesvizky R et al. Integrated safety profile of single-agent carfilzomib: experience from 526 patients enrolled in 4 phase II clinical studies. Haematologica 2013; 98: 1753–1761.
Korde N, Zingone A, Kwok M, Manasanch EE, Costello R, Zuchlinski D et al. Phase II clinical and correlative study of carfilzomib, lenalidomide, and dexamethasone (CRd) in newly diagnosed multiple myeloma (MM) patients. ASH Annu Meet Abstr 2012; 120: 732.
Niesvizky R, Martin TG, Bensinger WI, Alsina M, Siegel DS, Kunkel LA et al. Phase Ib dose-escalation study (PX-171-006) of carfilzomib, lenalidomide, and low-dose dexamethasone in relapsed or progressive multiple myeloma. Clin Cancer Res 2013; 19: 2248–2256.
Badros AZ, Papadopoulos KP, Zojwalla N, Lee JR, Siegel DSA . Phase 1b study of 30-minute infusion carfilzomib 20/45 and 20/56 mg/m2 plus 40 mg weekly dexamethasone in patients with relapsed and/or refractory (R/R) multiple myeloma. ASH Annu Meet Abstr 2012; 120: 4036.
Shah JJ, Stadtmauer EA, Abonour R, Cohen AD, Bensinger WI, Gasparetto C et al. A multi-center phase I/II trial of carfilzomib and pomalidomide with dexamethasone (Car-Pom-d) in patients with relapsed/refractory multiple myeloma. ASH Annu Meet Abstr 2012; 120: 74.
Touzeau C, Kolb B, Hulin C, Caillot D, Benboubker L, Tiab M et al. Effect of CMP, carfilzomib (CFZ) plus melphalan-prednisone (MP), on response rates in elderly patients (pts) with newly diagnosed multiple myeloma (NDMM): results of a phase (Ph) I/II trial. J Clin Oncol 2013; 31: 8513.
Palumbo A, Bringhen S, Villani O, Siniscalchi A, Russo E, Uccello G et al. Carfilzomib, cyclophosphamide and dexamethasone (CCd) for newly diagnosed multiple myeloma (MM) patients. ASH Annu Meet Abstr 2012; 120: 730.
Sonneveld P, Asselbergs E, Zweegman S, Van der Holt B, Kersten MJ, Vellenga E et al. Carfilzomib combined with thalidomide and dexamethasone (CTD) is an highly effective induction and consolidation treatment in newly diagnosed patients with multiple myeloma (MM) who are transplant candidate. ASH Annu Meet Abstr 2012; 120: 333.
Mikhael JR, Reeder CB, Libby ENI, Costa LJ, Bergsagel PL, Buadi F et al. Results from the phase II dose expansion of cyclophosphamide, carfilzomib, thalidomide and dexamethasone (CYCLONE) in patients with newly diagnosed multiple myeloma. ASH Annu Meet Abstr 2012; 120: 445.
Richardson PG, Weller E, Jagannath S, Avigan DE, Alsina M, Schlossman RL et al. Multicenter, phase I, dose-escalation trial of lenalidomide plus bortezomib for relapsed and relapsed/refractory multiple myeloma. J Clin Oncol 2009; 27: 5713–5719.
Richardson PG, Jagannath S, Jakubowiak AJ, Lonial S, Raje N, Alsina M et al. Phase II trial of lenalidomide, bortezomib, and dexamethasone in patients (pts) with relapsed and relapsed/refractory multiple Myeloma (MM): updated efficacy and safety data after >2 years of follow-up. ASH Annu Meet Abstr 2010; 116: 3049.
Groll M, Kim KB, Kairies N, Huber RCC . Crystal structure of epoxomicin:20S proteasome reveals a molecular basis for selectivity of α′,β′-epoxyketone proteasome inhibitors. J Am Chem Soc 2000; 122: 1237–1238.
Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN et al. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res 2007; 67: 6383–6391.
Ri M, Iida S, Nakashima T, Miyazaki H, Mori F, Ito A et al. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010; 24: 1506–1512.
Jagannath S, Barlogie B, Berenson JR, Siegel DS, Irwin D, Richardson PG et al. Updated survival analyses after prolonged follow-up of the phase 2, multicenter CREST study of bortezomib in relapsed or refractory multiple myeloma. Br J Haematol 2008; 143: 537–540.
Bringhen S, Larocca A, Rossi D, Cavalli M, Genuardi M, Ria R et al. Efficacy and safety of once-weekly bortezomib in multiple myeloma patients. Blood 2010; 116: 4745–4753.
Acknowledgements
We thank all of the patients, families and caregivers who contributed and participated in this study. We acknowledge Liv Thulin and Melissa Kalbarczyk for invaluable assistance with data management and entry. We also acknowledge A Peter Morello III (Onyx Pharmaceuticals Inc) for providing medical writing and editorial assistance, which was funded by Onyx Pharmaceuticals Inc. This study was funded by Onyx Pharmaceuticals Inc.
Author contributions
JRB designed and performed research, analyzed data and wrote the paper; JDH designed research, analyzed data and wrote the paper. OY designed research. RD, DP-D, RVB, AB, LS, DG, SE, YN, RAS and RAV performed research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
JRB and RAS are consultants and on the speakers bureau for Onyx Pharmaceuticals Inc., and receive research funding from Onyx Pharmaceuticals Inc. RVB is on the speakers bureau for Onyx Pharmaceuticals Inc. The remaining authors declare no conflict of interest.
Additional information
Presented in abstract form at the 43rd annual meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 2 June 2013.
Supplementary Information accompanies this paper on the Leukemia website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Berenson, J., Hilger, J., Yellin, O. et al. Replacement of bortezomib with carfilzomib for multiple myeloma patients progressing from bortezomib combination therapy. Leukemia 28, 1529–1536 (2014). https://doi.org/10.1038/leu.2014.27
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2014.27
Keywords
This article is cited by
-
Proteasome Inhibitor-Related Cardiotoxicity: Mechanisms, Diagnosis, and Management
Current Oncology Reports (2020)
-
Carfilzomib–lenalidomide–dexamethasone vs lenalidomide–dexamethasone in relapsed multiple myeloma by previous treatment
Blood Cancer Journal (2017)
-
Outcomes of multiple myeloma patients receiving bortezomib, lenalidomide, and carfilzomib
Annals of Hematology (2017)
-
Pooled analysis of the reports of carfilzomib, panobinostat, and elotuzumab combinations in patients with refractory/relapsed multiple myeloma
Journal of Hematology & Oncology (2016)
-
Tumor lysis syndrome in the era of novel and targeted agents in patients with hematologic malignancies: a systematic review
Annals of Hematology (2016)
