
Abstract
The majority of patients with BCR-ABL1-negative myeloproliferative neoplasms (MPN) harbor mutations in JAK2 or MPL, which lead to constitutive activation of the JAK/STAT, PI3K and ERK signaling pathways. JAK inhibitors by themselves are inadequate in producing selective clonal suppression in MPN and are associated with hematopoietic toxicities. MK-2206 is a potent allosteric AKT inhibitor that was well tolerated, including no evidence of myelosuppression, in a phase I study of solid tumors. Herein, we show that inhibition of PI3K/AKT signaling by MK-2206 affected the growth of both JAK2V617F- or MPLW515L-expressing cells via reduced phosphorylation of AKT and inhibition of its downstream signaling molecules. Moreover, we demonstrate that MK-2206 synergizes with ruxolitinib in suppressing the growth of JAK2V617F-mutant SET2 cells. Importantly, MK-2206 suppressed colony formation from hematopoietic progenitor cells in patients with primary myelofibrosis and alleviated hepatosplenomegaly and reduced megakaryocyte burden in the bone marrows, livers and spleens of mice with MPLW515L-induced MPN. Together, these findings establish AKT as a rational therapeutic target in the MPNs.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
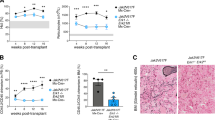
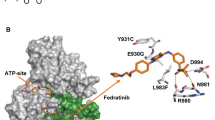

References
Ma X, Vanasse G, Cartmel B, Wang Y, Selinger HA . Prevalence of polycythemia vera and essential thrombocythemia. Am J Hematol 2008; 83: 359–362.
Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005; 7: 387–397.
Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JR et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779–1790.
James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005; 434: 1144–1148.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054–1061.
Zhao R, Xing S, Li Z, Fu X, Li Q, Krantz SB et al. Identification of an acquired JAK2 mutation in polycythemia vera. J Biol Chem 2005; 280: 22788–22792.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937–951.
Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010; 363: 1117–1127.
Tefferi A . Mutations galore in myeloproliferative neoplasms: would the real Spartacus please stand up? Leukemia 2011; 25: 1059–1063.
Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med 2006; 3: e270.
Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008; 112: 141–149.
Wernig G, Mercher T, Okabe R, Levine RL, Lee BH, Gilliland DG . Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 2006; 107: 4274–4281.
Zaleskas VM, Krause DS, Lazarides K, Patel N, Hu Y, Li S et al. Molecular pathogenesis and therapy of polycythemia induced in mice by JAK2 V617F. PLoS One 2006; 1: e18.
Lacout C, Pisani DF, Tulliez M, Gachelin FM, Vainchenker W, Villeval JL . JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006; 108: 1652–1660.
Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F et al. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer Cell 2010; 17: 584–596.
Pardanani A, Guglielmelli P, Lasho TL, Pancrazzi A, Finke CM, Vannucchi AM et al. Primary myelofibrosis with or without mutant MPL: comparison of survival and clinical features involving 603 patients. Leukemia 2011; 25: 1834–1839.
Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A et al. Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289–2301.
Carbuccia N, Murati A, Trouplin V, Brecqueville M, Adelaide J, Rey J et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009; 23: 2183–2186.
Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013; e-pub ahead of print 17 May 2013; doi:10.1038/leu.2013.119.
Lasho TL, Finke CM, Hanson CA, Jimma T, Knudson RA, Ketterling RP et al. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia 2012; 26: 1135–1137.
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med 2012; 366: 799–807.
Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol 2011; 29: 789–796.
Pardanani A, Laborde RR, Lasho TL, Finke C, Begna K, Al-Kali A et al. Safety and efficacy of CYT387, a JAK1 and JAK2 inhibitor, in myelofibrosis. Leukemia 2013; 27: 1322–1327.
Tefferi A . JAK inhibitors for myeloproliferative neoplasms: clarifying facts from myths. Blood 2012; 119: 2721–2730.
Ugo V, Marzac C, Teyssandier I, Larbret F, Lecluse Y, Debili N et al. Multiple signaling pathways are involved in erythropoietin-independent differentiation of erythroid progenitors in polycythemia vera. Exp Hematol 2004; 32: 179–187.
Laubach JP, Fu P, Jiang X, Salter KH, Potti A, Arcasoy MO . Polycythemia vera erythroid precursors exhibit increased proliferation and apoptosis resistance associated with abnormal RAS and PI3K pathway activation. Exp Hematol 2009; 37: 1411–1422.
Grimwade LF, Happerfield L, Tristram C, McIntosh G, Rees M, Bench AJ et al. Phospho-STAT5 and phospho-Akt expression in chronic myeloproliferative neoplasms. Br J Haematol 2009; 147: 495–506.
Wang Y, Fiskus W, Chong DG, Buckley KM, Natarajan K, Rao R et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009; 114: 5024–5033.
Akada H, Yan D, Zou H, Fiering S, Hutchison RE, Mohi MG . Conditional expression of heterozygous or homozygous Jak2V617F from its endogenous promoter induces a polycythemia vera-like disease. Blood 2010; 115: 3589–3597.
Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest 2008; 118: 3038–3050.
Bogani C, Bartalucci N, Martinelli S, Tozzi L, Guglielmelli P, Bosi A et al. mTOR inhibitors alone and in combination with JAK2 inhibitors effectively inhibit cells of myeloproliferative neoplasms. PLoS One 2013; 8: e54826.
Fiskus W, Verstovsek S, Manshouri T, Smith JE, Peth K, Abhyankar S et al. Dual PI3K/AKT/mTOR inhibitor BEZ235 synergistically enhances the activity of JAK2 inhibitor against cultured and primary human myeloproliferative neoplasm cells. Mol Cancer Ther 2013; 12: 577–588.
Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res 2010; 16: 5424–5435.
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 2010; 70: 8097–8107.
Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012; 30: 282–290.
Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol 2011; 29: 4688–4695.
Uozumi K, Otsuka M, Ohno N, Moriyama T, Suzuki S, Shimotakahara S et al. Establishment and characterization of a new human megakaryoblastic cell line (SET-2) that spontaneously matures to megakaryocytes and produces platelet-like particles. Leukemia 2000; 14: 142–152.
Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 2010; 9: 1956–1967.
Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK et al. Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood 2011; 118: 1264–1273.
Wernig G, Kharas MG, Mullally A, Leeman DS, Okabe R, George T et al. EXEL-8232, a small-molecule JAK2 inhibitor, effectively treats thrombocytosis and extramedullary hematopoiesis in a murine model of myeloproliferative neoplasm induced by MPLW515L. Leukemia 2012; 26: 720–727.
Koppikar P, Abdel-Wahab O, Hedvat C, Marubayashi S, Patel J, Goel A et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood 2010; 115: 2919–2927.
Tyner JW, Bumm TG, Deininger J, Wood L, Aichberger KJ, Loriaux MM et al. CYT387, a novel JAK2 inhibitor, induces hematologic responses and normalizes inflammatory cytokines in murine myeloproliferative neoplasms. Blood 2010; 115: 5232–5240.
Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G et al. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia 2007; 21: 1658–1668.
Santos FP, Kantarjian HM, Jain N, Manshouri T, Thomas DA, Garcia-Manero G et al. Phase 2 study of CEP-701, an orally available JAK2 inhibitor, in patients with primary or post-polycythemia vera/essential thrombocythemia myelofibrosis. Blood 2010; 115: 1131–1136.
Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998; 93: 385–395.
Begna KH, Mesa RA, Pardanani A, Hogan WJ, Litzow MR, McClure RF et al. A phase-2 trial of low-dose pomalidomide in myelofibrosis. Leukemia 2011; 25: 301–304.
Guerini V, Barbui V, Spinelli O, Salvi A, Dellacasa C, Carobbio A et al. The histone deacetylase inhibitor ITF2357 selectively targets cells bearing mutated JAK2(V617F). Leukemia 2008; 22: 740–747.
Rambaldi A, Dellacasa CM, Finazzi G, Carobbio A, Ferrari ML, Guglielmelli P et al. A pilot study of the Histone-Deacetylase inhibitor Givinostat in patients with JAK2V617F positive chronic myeloproliferative neoplasms. Br J Haematol 2010; 150: 446–455.
Mascarenhas J, Lu M, Li T, Petersen B, Hochman T, Najfeld V et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br J Haematol 2013; 161: 68–75.
Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest 2010; 120: 3578–3593.
Fiskus W, Verstovsek S, Manshouri T, Rao R, Balusu R, Venkannagari S et al. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res 2011; 17: 7347–7358.
Guglielmelli P, Barosi G, Rambaldi A, Marchioli R, Masciulli A, Tozzi L et al. Safety and efficacy of everolimus, a mTOR inhibitor, as single agent in a phase 1/2 study in patients with myelofibrosis. Blood 2011; 118: 2069–2076.
Engelman JA . Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9: 550–562.
Simioni C, Neri L, Tabellini G, Ricci F, Bressanin D, Chiarini F et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 2012; 26: 2336–2342.
Cheng Y, Zhang Y, Zhang L, Ren X, Huber-Keener KJ, Liu X et al. MK-2206, a novel allosteric inhibitor of Akt, synergizes with gefitinib against malignant glioma via modulating both autophagy and apoptosis. Mol Cancer Ther 2012; 11: 154–164.
Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One 2010; 5: e14124.
Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007; 110: 986–993.
Stankiewicz MJ, Crispino JD . AKT collaborates with ERG and Gata1s to dysregulate megakaryopoiesis and promote AMKL. Leukemia 2013; 27: 1339–1347.
Acknowledgements
We thank Jonathan Licht and Lou Dore for their helpful advice and critical reading of the manuscript. We also thank Merck for supplying MK-2206. This work was supported in part by grants from the NIH (CA101774 to JDC) and the Leukemia and Lymphoma Society, the Samuel Waxman Cancer Research Foundation, National Natural Science Foundation of China (Grant No. 30700412 and 81070406 to Z Huang). IK was supported by a T32 Grant to Northwestern University. IK is a recipient of the American Society of Hematology Translational Research Training in Hematology (TRTH) Award.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Author Contributions
IK, ZH, QW, LG, BG performed the experiments, analyzed the data and wrote the manuscript. PK, LD, MJS and RS assisted with experiments and animal studies. SG analyzed the data and assisted with the manuscript. CF, TL, QW, MS, AP, BS, JA and AT assisted with patient specimen experiments. RL, AT and JC analyzed the data and wrote the manuscript.
Supplementary Information accompanies this paper on the Leukemia website
Supplementary information
Rights and permissions
About this article
Cite this article
Khan, I., Huang, Z., Wen, Q. et al. AKT is a therapeutic target in myeloproliferative neoplasms. Leukemia 27, 1882–1890 (2013). https://doi.org/10.1038/leu.2013.167
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2013.167
Keywords
This article is cited by
-
JAK2 inhibitors for the treatment of Philadelphia-negative myeloproliferative neoplasms: current status and future directions
Molecular Diversity (2023)
-
Combined treatment with ruxolitinib and MK-2206 inhibits the JAK2/STAT5 and PI3K/AKT pathways via apoptosis in MDA-MB-231 breast cancer cell line
Molecular Biology Reports (2023)
-
JAK2 inhibitor persistence in MPN: uncovering a central role of ERK activation
Blood Cancer Journal (2022)
-
MAPK14 over-expression is a transcriptomic feature of polycythemia vera and correlates with adverse clinical outcomes
Journal of Translational Medicine (2021)
-
The Next Generation of JAK Inhibitors: an Update on Fedratinib, Momelotonib, and Pacritinib
Current Hematologic Malignancy Reports (2020)
