
Abstract
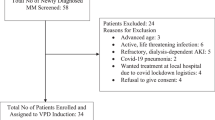
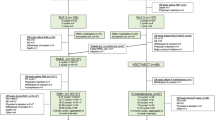
This multicenter phase II trial evaluated the safety and efficacy of lenalidomide−prednisone (RP) induction, followed by lenalidomide−melphalan−prednisone (MPR) consolidation and RP maintenance in elderly unfit newly diagnosed myeloma patients. Patients received four 28-day RP induction courses (lenalidomide 25 mg/day on days 1–21 and prednisone 50 mg three times/week), followed by six 28-day MPR consolidation cycles (melphalan 2 mg, prednisone 50 mg three times/week and lenalidomide 10–15 mg/day on days 1–21), and maintenance with lenalidomide (10 mg/day on days 1–21 every 28 days) plus prednisone (25 mg three times/week). Forty-six patients were enrolled. Median age was 75 years, 59% of patients had at least one comorbidity and 35% at least two. Partial response rate was 80%, including 29% very good partial response. Median time to progression was 19.6 months, median progression-free survival was 18.4 months and 2-year overall survival was 80%. At the tolerated consolidation dose (melphalan 25 mg/month and lenalidomide 10 mg/day), the most frequent grade 3 adverse events were neutropenia (36.4%), anemia (12.1%), cutaneous reactions (18.2%) and infections (12.1%). Grade 4 neutropenia occurred in 12.1% of patients. In conclusion, RP induction followed by MPR consolidation and RP maintenance showed a manageable safety profile, and reduced the risk of severe hematological toxicity in unfit elderly myeloma patients.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Altekruse SF, Kosary CL, Krapcho M, Neyman N, Aminou R, Waldron W et al. (eds). SEER Cancer Statistics Review, 1975-2007, National Cancer Institute: Bethesda, MD (http://seer.cancer.gov/csr/1975_2007/; based on November 2009 SEER data submission, posted to the SEER website, 2010.
Kristinsson SY, Landgren O, Dickman PW, Derolf AR, Bjorkholm M . Patterns of survival in multiple myeloma: a population-based study of patients diagnosed in Sweden from 1973 to 2003. J Clin Oncol 2007; 25: 1993–1999.
Brenner H, Gondos A, Pulte D . Recent major improvements in long-term survival of younger patients with multiple myeloma. Blood 2008; 111: 2521–2526.
Schaapveld M, Visser O, Siesling S, Schaar CG, Zweegman S, Vellenga E . Improved survival among younger but not among older patients with multiple myeloma in the Netherlands, a population- based study since 1989. Eur J Cancer 2010; 46: 160–169.
Kumar SK, Rajkumar SV, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood 2008; 111: 2516–2520.
Palumbo A, Bringhen S, Ludwig H, Dimopoulos MA, Bladé J, Mateos MV et al. Personalized therapy in multiple myeloma according to patients age and vulnerability: a report of the European Multiple Myeloma Network (EMN). Blood 2011; 118: 4519–4529.
Fayers PM, Palumbo A, Hulin C, Waage A, Wijermans P, Beksaç M et al. Thalidomide for previously untreated elderly patients with multiple myeloma: meta-analysis of 1685 individual patient data from six randomized clinical trials. Blood 2011; 118: 1239–1247.
San Miguel JF, Schlag R, Khuageva NK, Dimopoulos MA, Shpilberg O, Kropff M et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med 2008; 359: 906–917.
Palumbo A, Anderson K . Multiple myeloma. N Engl J Med 2011; 364: 1046–1060.
Chanan-Khan AA, Cheson BD . Lenalidomide for the treatment of B-cell malignancies. J Clin Oncol 2008; 26: 1544–1552.
Rajkumar SV, Hayman SR, Lacy MQ, Dispenzieri A, Geyer SM, Kabat B et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005; 106: 4050–4053.
Gay F, Haymann SR, Lacy MQ, Buadi F, Gertz MA, Kumar S et al. Lenalidomide plus dexamethasone vs. thalidomide plus dexamethasone in newly diagnosed multiple myeloma: a comparative analysis of 411 patients. Blood 2010; 115: 1343–1350.
Rajkumar SV, Jacobus S, Callander NS, Fonseca R, Vesole DH, Williams ME et al. Lenalidomide plus high-dose dexamethasone versus lenalidomide plus low-dose dexamethasone as initial therapy for newly diagnosed multiple myeloma: an open-label randomised controlled trial. Lancet Oncol 2010; 11: 29–37.
Palumbo A, Falco P, Corradini P, Falcone A, Di Raimondo F, Giuliani N et al. Melphalan, prednisone, and lenalidomide treatment for newly diagnosed myeloma: a report from the GIMEMA--Italian Multiple Myeloma Network. J Clin Oncol 2007; 25: 4459–4465.
Palumbo A, Falco P, Falcone A, Benevolo G, Canepa L, Gay F et al. Melphalan, prednisone and lenalidomide for newly diagnosed myeloma: kinetics of neutropenia and thrombocytopenia and time-to-event results. Clin Lymphoma Myeloma 2009; 9: 145–150.
Palumbo A, Hajek R, Delforge M . Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med 2012; 366: 1759–1769.
Bryant J, Day R . Incorporating toxicity considerations into the design of two-stage phase II clinical trials. Biometrics 1995; 51: 1372–1383.
Durie BMG, Harousseau JL, Miguel JS, Bladé J, Barlogie B, Anderson K et al. International uniform criteria for multiple myeloma. Leukemia 2006; 20: 1467–1473.
Cancer Therapy Evaluation Program. Common Terminology Criteria For Adverse Events, version 3.0, DCTD, NCI, NIH, DHHS 31 march 2003 http://ctep.cancer.gov (accessed 12 December 2003).
Kaplan EL, Meier P . Non parametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–481.
Hutchins LF, Unger JM, Crowley JJ, Coltman CA, Albain KS . Underrepresentation of patients 65 years of age or older in cancer-treatment trials. N Engl J Med 1999; 341: 2061–2067.
Gay F, Rajkumar V, Falco P, Kumar S, Dispenzieri A, Petrucci MT et al. Lenalidomide plus dexamethasone vs. lenalidomide plus melphalan and prednisone: a retrospective study in newly diagnosed multiple myeloma. Eur J Haemat 2010; 85: 200–208.
Gay F, Haymann SR, Buadi F, Detweiler-Short K, Lacy M, Kumar S et al. Safety and efficacy of lenalidomide-dexamethasone in elderly newly diagnosed multiple myeloma patients aged 70 and older. Haematologica 2010; 95: 153 (abstract 0377).
Harrison SJ, Khot AS, Tsin T, Hsu A, Chen K, Loudovaris M et al. Low dose lenalidomide and dexamethasone induction followed by autologous transplantation in untreated patients with myeloma is associated with high response rates and preservation of CD8, but not CD4 or NK cellular immunity. Blood 2011; 118: 1862 (abstract 1862).
Niesvizky R, Flinn IW, Rifkin R, Gabrail N, Charu V, Clowney B et al. Efficacy and safety of three bortezomib-based combinations in elderly, newly diagnosed multiple myeloma patients: results from all randomized patients in the community-based, phase 3b UPFRONT study. Blood 2011; 118: 1864 (abstract 478).
Kapoor P, Kumar S, Fonseca R, Lacy MQ, Witzig TE, Hayman SR et al. Impact of risk stratification on outcome among patients with multiple myeloma receiving initial therapy with lenalidomide and dexamethasone. Blood 2009; 114: 518–521.
Dimopoulos MA, Kastritis E, Christoulas D, Migkou M, Gavriatopoulou M, Gkotzamanidou M et al. Treatment of patients with relapsed/refractory multiple myeloma with lenalidomide and dexamethasone with or without bortezomib: prospective evaluation of the impact of cytogenetic abnormalities and of previous therapies. Leukemia 2010; 24: 1769–1778.
Kumar SK, Lacy MQ, Hayman SR, Stewart K, Buadi FK, Allred J et al. Lenalidomide, cyclophosphamide and dexamethasone (CRd) for newly diagnosed multiple myeloma: results from a phase 2 trial. Am J Hematol 2011; 86: 640–645.
Acknowledgements
We thank the patients who took part in the study, the Rete Oncologica del Piemonte e della Valle d’Aosta, the nurses Manuela Grasso and Loredana Puccio, the data managers Federica Leotta and Elena Tigano and the editorial assistant Giorgio Schirripa.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PF has received honoraria from Celgene, Janssen-Cilag and Novartis; FC has received honoraria from Celgene, Janssen-Cilag, Onyx, and served on the advisory committee for Celgene; AL has received honoraria from Celgene and Janssen-Cilag; TG has received honoraria from Celgene and Janssen-Cilag; PM has received honoraria from Celgene; MB has received research funding from and served on the advisory board for Celgene and Janssen-Cilag; AP has received honoraria from Celgene, Janssen-Cilag, Bristol-Myers Squibb, Millenium, Merck, Onyx, and served on the advisory board for Celgene and Janssen-Cilag. The remaining authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Falco, P., Cavallo, F., Larocca, A. et al. Lenalidomide−prednisone induction followed by lenalidomide−melphalan−prednisone consolidation and lenalidomide−prednisone maintenance in newly diagnosed elderly unfit myeloma patients. Leukemia 27, 695–701 (2013). https://doi.org/10.1038/leu.2012.271
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2012.271
Keywords
This article is cited by
-
History of autoimmune disease is associated with impaired survival in multiple myeloma and monoclonal gammopathy of undetermined significance: a population-based study
Annals of Hematology (2017)
-
Management of Elderly Patients with Plasma Cell Myeloma
Drugs & Aging (2015)
-
Lenalidomide in relapsed and refractory multiple myeloma disease: feasibility and benefits of long-term treatment
Annals of Hematology (2014)
-
Therapiekonzepte bei älteren Patienten mit multiplem Myelom
Der Onkologe (2014)
-
New Approaches to Management of Multiple Myeloma
Current Treatment Options in Oncology (2014)
