
Abstract
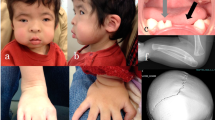
Alveolar capillary dysplasia (ACD) is a rare and lethal cause of hypoxic respiratory failure in the neonate. Here we describe a term neonate with ACD that was found with a previously unreported p.Arg86Pro mutation in the FOXF1 (Forkhead Box-F1) gene and coexisting congenital anomalies, including colobomas of the iris and hemihyperplasia. This unique clinical presentation may indicate a novel, yet unconfirmed disease association for mutations in the FOXF1 gene. Rapid mutation analysis in FOXF1 may provide noninvasive early confirmation of ACD in neonates with respiratory failure and can aid in clinical decision making.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Bishop NB, Stankiewicz P, Steinhorn RH . Alveolar capillary dysplasia. Am J Respir Crit Care Med 2011; 184: 172–179.
Galambos C, Sims-Lucas S, Abman SH . Three-dimensional reconstruction identifies misaligned pulmonary veins as intrapulmonary shunt vessels in alveolar capillary dysplasia. J Pediatr 2014; 164 (1): 192–195.
Sen P, Thakur N, Stockton DW, Langston C, Bejani BA . Expanding the phenotype of alveolar capillary dysplasia (ACD). J Pediatr 2004; 145: 646–651.
Boggs S, Harris MC, Hoffman DJ, Goel R, McDonald-McGinn D, Langston C et al. Misalignment of pulmonary veins with alveolar capillary dysplasia: Affected siblings and variable phenotypic expression. J Pediatr 1994; 124: 125–128.
Yu S, Shao L, Killbride H, Zwick L . Haploinsufficiencies of FOXF1 and FOXC2 genes associated with lethal alveolar capillary dysplasia and congenital heart disease. Am J Med Genet 2010; 152A: 1257–1262.
Stankiewicz P, Sen P, Bhatt SS, Storer M, Xia Z, Bejjani BA et al. Genomic and genic deletions of the FOX gene cluster on 16q24.1 and inactivating mutations of FOXF1 cause alveolar capillary dysplasia and other malformations. Am J Hum Genet 2009; 84: 780–791.
Sen P, Yang Y, Navarro C, Silva I, Szafranski P, Kolodziejska KE et al. Novel FOXF1 mutations in sporadic and familial cases of alveolar capillary dysplasia with misaligned pulmonary veins imply a role for its dna binding domain. Hum Mutat 2013; 32: 801–811.
Sen P, Gerychova R, Janku P, Jezova M, Valaskova I, Navarro C et al. A familial case of alveolar capillary dysplasia with misalignment of pulmonary veins supports paternal imprinting of FOXF1 in human. Euro J Hum Genet 2013; 21: 474–477.
Pasutto F, Sticht H, Hammersen G, Gilleseen-Kaesbach G, FitzPatrick DR, Nurnberg G et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia. alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am J Hum Genet 2007; 80: 550–560.
Acknowledgements
We thank the family of this patient for consenting to this publication and Baylor College of Medicine’s Medical Genetics Laboratories for expediting the clinical confirmation of the genotype for this patient. We also thank the DNA-sequencing laboratory of the HMGC at the Medical College of Wisconsin for providing rapid DNA sequencing on a research basis, which provided concurrent results in <1 week.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Geddes, G., Dimmock, D., Hehir, D. et al. A novel FOXF1 mutation associated with alveolar capillary dysplasia and coexisting colobomas and hemihyperplasia. J Perinatol 35, 155–157 (2015). https://doi.org/10.1038/jp.2014.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jp.2014.187
