
Abstract
Tandem mass screening has recently been started in Japan, but genetic screening has yet to be widely performed in neonates and many unexpected deaths are still being reported. We previously reported two cases of sudden infant death that may have been prevented had newborn screening been performed. In this study, we retrospectively reviewed 71 cases of sudden infant death for 66 arrhythmia- and 63 metabolic disease-related genes to identify how many cases of sudden infant death may have been prevented had mass screening been performed. Next-generation sequencing revealed that six cases had arrhythmia-related gene variants and five cases had metabolic disease-related gene variants. Had genetic screening been performed in addition to biochemical and physiological screening during the neonatal period to identify those at risk of arrhythmia or metabolic disease, these infants could have been diagnosed and treated, preventing their deaths. As such, screening of newborns may prevent sudden infant death.
Similar content being viewed by others


Introduction
Sudden unexpected death (SUD) in infancy is described as the sudden death of healthy infants within their first year of life, the cause of which is unknown. It includes deaths involving accidents, abuse, congenital diseases, inflammatory diseases and so on. If a complete examination, including an autopsy, cannot explain the exact cause of death, the case is diagnosed as sudden infant death syndrome. Although the number of sudden infant death syndrome cases has gradually decreased in Japan due to the ‘back to sleep’ or ‘safe to sleep’ campaign,1, 2 annually several hundreds of children still die suddenly and unexpectedly before or around the age of one in Japan.
Congenital diseases, including metabolic diseases related to fatty acid oxidation and ketone body metabolism, and genetic arrhythmia, are linked to SUD.3, 4, 5, 6, 7, 8, 9, 10 These diseases are generally asymptomatic in daily life, but may become symptomatic if left unnoticed and untreated. Some fatty acid oxidation disorders can be treated by avoiding fasting and providing glucose supplementation. Delaying diagnosis by even a few days can lead to permanent mental retardation, coma or sudden death. Genetic arrhythmia is also preventable with medical intervention. The diagnosis of these diseases before they become symptomatic may enable intervention that can prevent disease onset. As such, the early detection, diagnosis and treatment of these diseases at the asymptomatic phase are essential; this highlights the importance of newborn screening.
Tandem mass screening started in Japan in the 1990s, and is now available nationwide.11, 12 It can detect more than 20 congenital metabolic diseases in newborns within the first few days of life. Neonatal electrocardiographic screening has also been started and it can identify asymptomatic long QT patients.13 We previously reported two cases of SUD in infancy, whose causes of death were determined to be carnitine palmitoyltransferase (CPT) II deficiency.14, 15 These patients appeared healthy and the CPT II deficiency was not noticed before their deaths. If screening had been performed before their deaths, they could have been treated and saved.
In this study, we retrospectively reviewed 71 cases of SUD from a genetic viewpoint for arrhythmia and metabolic disease to reveal how many cases of sudden infant death may have been prevented had genetic screening been performed.
Materials and methods
Case collection
According to the selection criteria (1: died before or around the age of 1 year; 2: informed consent and/or approval from an ethics committee was obtained; 3: DNA samples were available; 4: no involvement of a congenital abnormality, infection, trauma, murder, or abuse.), a total of 71 cases of SUD occurring in infants aged under or around 1 year (35 males, 36 females; age range: 1 day to 1 year and 5 months) were selected (Supplementary Table 1). A comprehensive forensic investigation, including a thorough examination of the death scene, a review of the clinical history, and performance of an autopsy that included macroscopic and microscopic examinations and a toxicology examination, were performed in all cases. Phenylketonuria, maple syrup urine disease, homocystinuria, galactosemia, congenital adrenal hyperplasia or congenital hypothyroidism had not been detected in these cases using conventional Guthrie mass screening.
Extraction of genomic DNA and genetic analysis
Genomic DNA was isolated from blood leukocytes with the QIAamp DNA Blood Mini Kit (Qiagen, Tokyo, Japan) in accordance with the manufacturer’s standard methods. A TruSight One sequencing panel (Illumina, San Diego, CA, USA) was used and the sequencing was performed on an Illumina MiSeq (Illumina).
Targeted genes and filtering steps
The sequencing reads were mapped to the hg19 human reference genome sequence using Variant Studio (version 2.2.3) software (Illumina). From the 4813 TruSight One panel targeting genes, we extracted sequence information in the 129 genes for the inherited arrhythmia, arrhythmogenic cardiomyopathy and metabolic diseases; 19 BrS genes, 15 LQTS genes, 6 SQTS genes, 6 PCCD genes, 5 CPVT genes, 5 ARVC genes, other 24 cardiac genes and 63 inherited metabolic disease genes such as fatty acid oxidation, amino acid and organic acid disorders. Gene list and the criteria of gene selection are provided in Tables 1 and 2.
Single-nucleotide variations causing non-synonymous substitutions, nonsense substitutions or located at the splice site, and insertions/deletions occurring in the coding regions were retrieved. The variants with a low Q30 score and a read depth below 30 were excluded. Copy number variation was not analyzed in this study. To identify putatively pathogenic variants, variants with allele frequencies equal to or <1% in East Asian ethnic subgroups were retained and listed using data from the dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP), the 1000 Genomes Project (http://www.1000genomes.org), the NHLBI Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS), the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org) and the Genome Cohort Study of Tohoku Medical Megabank Organization (ToMMo) (http://ijgvd.megabank.tohoku.ac.jp). Fastq files generated by MiSeq Control Software were mapped on the hg19 human reference genome sequence, sorted and duplication-removed by using NovoAlign (version 3.03.02) and NovoSort (version 1.04.03) software packages (Novocraft, Petaling Jaya, Malaysia). To prepare target regions for read depth counting, coding regions of target genes, including multiple transcripts, stored in the GENCODE basic version 19 table were extracted from the UCSC genome browser database using an in-house script and merged by using the BEDTools package (version 2.22.1).16, 17, 18, 19 Depth counting was performed by using the DepthOfCoverage tool of the Genome Analysis Toolkit version 3.7.20 In silico algorithms, SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), were used to predict whether the detected variants would affect the function of each protein.
Sanger sequencing
Sanger sequencing was performed to confirm the detected pathogenic variants, as previously described.14 The PCR primers were designed using Primer3 version 0.4.0 (http://bioinfo.ut.ee/primer3-0.4.0).
Ethics
Written informed consent was obtained from the parents or the use of the samples in this study was approved by the ethics committees of Nagasaki University Graduate School of Medicine and Fukuoka University. This study was performed in accordance with the Declaration of Helsinki.
Results
Target sequence of sudden death cases
On average, ~18 million total reads were produced and ~13 million reads mapped to the targeted region in each sample. The mean coverage of the coding sequence was 115.3±41 reads, with an overall average gene level coverage at ⩾20 reads of 94.3±0.04%.
Detected variants aligned to the target 129 genes
After the filtering steps, the total number of detected variants that were aligned to the 129 target genes was 185. On average, each case had 1.4±1 variants of the 66 arrhythmia-related genes and 1.2±1.2 variants of the 63 metabolic disease-related genes. The overall mean depth of each coding region was described in Supplementary Table 2.
Arrhythmia-related gene variants
Genetic arrhythmia
Most cases of genetic arrhythmia have autosomal dominant inheritance, and a familial history is not always present. Sporadic cases are always caused by de novo mutation and sometimes by germline mosaicism, but in some cases, either one of the parents may be a subclinical patient.
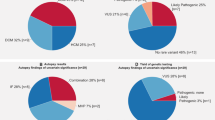
Among our 71 cases, 56 cases had at least one heterozygous amino acid change in an arrhythmia-related gene. Out of these 56 cases, 6 cases had at least one heterozygous amino acid change in an arrhythmia-related gene that had previously been reported to be an arrhythmia-related variant (Table 3). Novel or rare variants with uncertain significance were also detected in the arrhythmia-related genes (Supplementary Table 3).
Long QT syndrome-related variant
Case 10 had a R148W-KCNH2 variant. The KCNH2 gene encodes potassium voltage-gated channel subfamily H member 2. Mutation of this gene results in long QT syndrome, which causes ventricular arrhythmia and increases the risk of sudden death.21 It has been contested whether the R148W-KCNH2 variant is indeed a mutation10, 22, 23 or just a polymorphism.24, 25 Recently, Mechakra et al.26 revealed that the human ether-à-go-go-related gene (hERG) current was decreased when the hERG/R148W variant was co-expressed with the wild-type gene, indicating that the heterozygous R148W-KCNH2 variant causes long QT syndrome and is associated with Torsade de pointes, and that heterozygous carriers of the R148W-KCNH2 variant may be at risk of cardiac sudden death.
Case 68 had a A283V-KCNQ1 variant. The KCNQ1 gene encodes potassium voltage-gated channel subfamily Q member 1, and mutation of this gene causes long QT syndrome. This variant has not been reported before, but the A283G-KCNQ1 and A283T-KCNQ1 variants have been previously reported in long QT syndrome patients.27, 28
Case 25 had D85N-KCNE1 variant. The D85N-KCNE1 variant is a rare variant, but it has been reported to be disease-causing.29 In Xenopus oocytes, KCNQ1-encoded currents were reduced by about 50% by the D85N-KCNE1 variant.30 Nishio et al.29 also demonstrated that the D85N-KCNE1 variant significantly reduced wild-type KCNH2/KCNE1-encoded currents by about 30%.
Arrhythmogenic right ventricular dysplasia-related variant
Case 60 had a V158A-DSG2 variant. The DSG2 gene encodes a member of the desmoglein family and cadherin cell adhesion molecule superfamily of proteins, and mutation of this gene causes arrhythmogenic right ventricular dysplasia, familial, 10. This variant has not been reported before, but the V158G-DSG2 variant has been reported as non-pathogenic,31 although it has been detected in a family with arrhythmogenic right ventricular cardiomyopathy/dysplasia.32
Brugada syndrome-related variant
Case 16 had a F386C-SCN10A variant. The SCN10A gene encodes sodium voltage-gated channel alpha subunit 10, and mutation of this gene is related to Brugada syndrome.33, 34 The F386C-SCN10A variant has been reported as putative pathogenic mutation.35
Case 22 had a V110I-SCN3B variant. The SCN3B gene encodes sodium voltage-gated channel beta subunit 3 and is a modifier protein of the cardiac sodium channel complex. We reported that the V110I-SCN3B variant is a relatively common cause of SCN5A-negative Brugada syndrome in Japan, which results in a reduced sodium current because of the loss of cell surface expression of a pore-forming subunit of the cardiac sodium channel complex.36
Digenic variants
Case 43 had T4A-KCNE3 and A283V-KCNQ1 variants. The T4A-KCNE3 variant has previously been reported in a patient with Brugada syndrome, and its functional consequence was a gain of function of the transient outward potassium current, which may underlie the pathogenesis of Brugada-pattern electrocardiogram (ECG).37 In this case, the KCNE3 and KCNQ1 variants may have affected each other, leading to fatal arrhythmia.
Nonsense variants
There were three cases that had a stop-gain deletion with immature protein. Case 30 and 45 had deletions in the TRPM4 gene, and case 55 had a deletion in the KCNA5 gene. The TRPM4 gene encodes transient receptor potential cation channel subfamily M member 4 and is related to progressive familial heart block type I and progressive cardiac conduction disturbances,38, 39, 40 long QT syndrome and Brugada syndrome.41 The KCNA5 gene, which encodes potassium voltage-gated channel subfamily A member 5, is related to arterial fibrillation.42 However, according to the ExAC database, there was no significant difference between the expected number and the observed number of truncation variants, which means that both genes might have little effect on the disease.
Metabolic disease-related gene variants
Metabolic disease
Most inherited metabolic diseases have autosomal recessive inheritance. Homozygous amino acid changes or at least two heterozygous amino acid changes are necessary to cause metabolic disease, although a single heterozygous amino acid change with a large deletion or an intronic mutation may also cause disease.
Homozygous variants
During the filtering steps, 1 case out of the 71 cases had homozygous amino acid changes in the NAGS genes (Table 4).
Case 17 had a homozygous V241A-NAGS variant. The NAGS gene encodes N-acetylglutamate synthase, and mutation of this gene causes N-acetylglutamate synthase deficiency, a form of hyperammonemia.43, 44 As the variant was not detected in controls and the in silico algorithms predicted that it was deleterious and possibly damaging, it remains possible that this variant is related to the disease.
Heterozygous variants
Among the 71 cases, 49 cases had at least one heterozygous non-synonymous variant in the metabolic diseases-related genes; three of these cases had two variants in the same gene (in the ACADVL gene, HMGCL gene or CPT2 gene, respectively; Table 4).
Case 9 had G546R-ACADVL and C630S-ACADVL variants. Mutation of the ACADVL gene causes very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency. Although these two variants have not been reported to be common polymorphisms,45 this case may have had VLCAD deficiency.
Case 24 had T205S-HMGCL and M217V-HMGCL variants. Mutation of the HMGCL gene causes 3-hydroxy-3-methylglutaric aciduria.46 These two variantns have not been reported before, but the T205 amino acid is conserved among species and the D204N-HMGCL and G203E-HMGCL variants have respectively been reported in two patients.47, 48, 49 It is possible that the case had 3-hydroxy-3-methylglutaric aciduria.
Case 33 and 63 had a heterozygous F383Y-CPT2 variant and splice-site variant (c.233+2T>A). The F383Y-CPT2 variant causes a decrease in CPT II activity and has been reported in CPT II deficiency patients.14, 50, 51 Splice-site variants cause exon skipping and reduces CPT II activity.52, 53 These two cases may have had CPT II deficiency.
Three cases (cases 9, 17 and 63) also had arrhythmia-related variant, but all of them were variants with uncertain significance.
Discussion
Newborn screening
The first successful newborn screening was reported in the 1960s as the Guthrie qualitative bacterial inhibition test, which formed the basis for phenylketonuria programs.54 Millington et al. applied fast-atom bombardment tandem mass spectrometry to newborn screening of metabolic diseases in 1990,11, 55 and newborn screening has been continually improving since then.55, 56, 57, 58, 59, 60, 61, 62 In Japan, newborn screening with tandem mass spectrometry had only been performed in restricted regions as a pilot study,12 but it is now available nationwide.
Neonatal ECG screening has been started in some regions.13, 63 Yoshinaga et al.13 reported that among 4285 Japanese infants who were screened at the age of 1 month, four infants showed prolonged QT intervals. These patients were also genetically diagnosed. Two of them showed progressive prolongation of QT intervals, and medical intervention could be started. However, there have been many reports of fatal arrhythmia in SUD cases. Screening had not been performed in these fatal cases, and so the disease could not be diagnosed at an asymptomatic phase.
Genetic screening
The most common types of screening are tandem mass screening and neonatal ECG screening; they are performed biochemically and physiologically. However, no genetic screening is currently being widely performed in neonates. Although biochemical or physiological screening can identify many potential patients, there still remain some cases in which the diseases are missed. Edmondson et al.64 reported a 4-year-old CPT II deficiency patient whose disease was initially missed by newborn screening, demonstrating that not all cases of CPT II deficiency are being detected by the currently used newborn screening methods. Some potential long QT patients do not show extended QT intervals during the neonatal period because QT intervals change with aging; in contrast, a genetic variant remains the same in an individual throughout their lifetime, indicating the usefulness of genetic screening even in neonates despite the associated ethical issues and cost matters. More recently, next-generation sequencing has been developed and applied for the detection of genetic abnormalities in arrhythmia. This technique makes it possible to examine a much larger number of genes and exons in less time and at a lower cost, and it may enable genetic screening to become more feasible than it has been in the past.
Benefits and disadvantages
There are many benefits of genetic screening. First, in combination with biochemical and physiological screening, many potential patients may be detected before they reach the symptomatic phase. Second, genetic screening is useful for detecting diseases in which a genotype–phenotype correlation has been established. In such cases, appropriate therapeutic approaches could be applied on the basis of the identified genetic variants. Furthermore, the obtained genetic information could be useful for asymptomatic siblings and future pregnancies.
There are also some disadvantages to such approaches. Next-generation sequencing, such as whole-exome sequencing, might identify variants of unknown significance and coincidental pathogenic variants. Familial genetic counseling would also be necessary. Furthermore some undetected variants due to low-coverage regions or non-sequenced regions might be misdiagnosed. In this present study, some coding regions were not sequenced (Supplementary Table 2) and further analysis such as Sanger’s method is necessary for the future, but it is time consuming. In addition, the cost of next-generation sequencing is still rather expensive: it costed several hundred dollars per sample in this study.
Molecular autopsy
Postmortem genetic testing or molecular autopsy, including metabolic autopsy, has been commonly performed, but they targeted only one or a few genes. There are many genes that cause arrhythmia and metabolic disease, and thus, in many cases, the mutations were not detected. Recently, next-generation sequencing has also been used for postmortem genetic testing.65, 66, 67, 68 We also reported the first metabolic autopsy using next-generation sequencing.15
In this study, we analyzed 71 cases of SUD, most of which had not been undergone screening at the neonatal period. The number of targeted genes was 129, which covered most of the arrhythmia- and metabolic disease-related genes. The diseases related to these genes are preventable and worth screening. Among our cases, six cases (cases 10, 16, 22, 25, 43 and 68) had at least one heterozygous amino acid change in an arrhythmia-related gene that had previously been reported as an arrhythmia-related variant, 1 case (case 17) had homozygous amino acid changes in the NAGS gene, two cases (cases 9 and 24) had two heterozygous amino acid changes in the ACADVL and HMGCL genes, and two cases (cases 33 and 63) had a heterozygous amino acid change and splice-site variant in the CPT2 gene.
Arrhythmia diseases
There are two types of variants: crucial mutations and modifier substitutions. The variants of the KCNH2 and KCNQ1 genes are crucial mutations for long QT syndrome, whereas the variants of the SCN5A and SCN3B genes might be modifier substitutions for Brugada syndrome. Indeed, Probst et al.69 reported that SCN5A variants are not directly causal to the occurrence of Brugada phenotype in familial analyses. Therefore, those who have a crucial mutation should be investigated and provided intensive intervention, whereas familial analyses should be recommended to those with modifier substitutions.
Variants are also classified according to the disease onset period. Table 5 summarizes the data of previously reported patients with the variants detected in this study. The R148W-KCNH2 and A283T-KCNQ1 variants were detected during childhood, whereas the others were detected during adolescence. If large cohorts of patients are studied and an accurate genotype–phenotype correlation is established, the patients with a severe genotype could be provided intervention from an early period, and those with a mild genotype could be followed regularly.
Metabolic diseases
In this study, no definite diagnosis was made, although there were some cases in which metabolic diseases were suspected (cases 9, 17, 24, 33 and 63). Owing to the postmortem nature of the samples, enzymatic and biochemical analyses were not performed and definite diagnoses were not made. If genetic screening and tandem mass biochemical screening were combined, these patients could be filtered for potential diseases. Fatty acid oxidation disorders may be responsible for as much as 5% of SUD cases in infancy, and as we reported in our previous metabolic autopsy study, CPT II deficiency was the cause of SUD in two cases.14, 15 These deaths could have been prevented by newborn screening, demonstrating the necessity of early detection.
Limitations
There are some limitations in this study. First, diagnosis was only performed genetically, but actual diagnoses should be made in consideration of the clinical symptoms and examinations. In addition, not all patients with a genetic variant develop symptoms, and not all patients who are diagnosed with the diseases have a genetic abnormality. Second, this is a retrospective study, and not a prospective cohort study. This study focused on SUD in infancy and included only SUD cases in certain areas of Japan. It is also important to keep in mind that not all patients with these diseases die, and there are far more living patients suffering from the diseases than those who died. Third, segregation in families was not performed. It is valuable to clarify the significance of each variant. Familial analysis is needed for the future. Fourth, we filtered out many genes other than the 129 arrhythmia- and metabolic disease-related genes studied. There are undoubtedly many other candidate genes that can cause SUD. For example, variants in some structural genes such as sarcomere genes and desmosome genes may induce arrhythmias without cardiac alteration.
Conclusion
In this study, we diagnosed several supposed cases of arrhythmia (6/71 cases; 8.5%) and metabolic disease (5/71 cases; 7%) among the 71 SUD cases analyzed. In addition to the two previously reported cases of CPT II deficiency, we found that the diseases in a certain number of infants could have been diagnosed and treated had genetic screening been performed. Genetic newborn screening may enable the prevention of SUD in patients who would otherwise die.
References
Gibson, E., Dembofsky, C. A., Rubin, S. & Greenspan, J. S. Infant sleep position practices 2 years into the ‘back to sleep’ campaign. Clin. Pediatr. 39, 285–289 (2000).
Krous, H. F., Beckwith, J. B., Byard, R. W., Rognum, T. O., Bajanowski, T., Corey, T. et al. Sudden infant death syndrome and unclassified sudden infant deaths: a definitional and diagnostic approach. Pediatrics 114, 234–238 (2004).
Bennett, M. J. & Powell, S. Metabolic disease and sudden, unexpected death in infancy. Hum. Pathol. 25, 742–746 (1994).
Lundemose, J. B., Kolvraa, S., Gregersen, N., Christensen, E. & Gregersen, M. Fatty acid oxidation disorders as primary cause of sudden and unexpected death in infants and young children: an investigation performed on cultured fibroblasts from 79 children who died aged between 0-4 years. Mol. Pathol. 50, 212–217 (1997).
Boles, R. G., Buck, E. A., Blitzer, M. G., Platt, M. S., Cowan, T. M., Martin, S. K. et al. Retrospective biochemical screening of fatty acid oxidation disorders in postmortem livers of 418 cases of sudden death in the first year of life. J. Pediatr. 132, 924–933 (1998).
Chace, D. H., DiPerna, J. C., Mitchell, B. L., Sgroi, B., Hofman, L. F. & Naylor, E. W. Electrospray tandem mass spectrometry for analysis of acylcarnitines in dried postmortem blood specimens collected at autopsy from infants with unexplained cause of death. Clin. Chem. 47, 1166–1182 (2001).
Ackerman, M. J., Siu, B. L., Sturner, W. Q., Tester, D. J., Valdivia, C. R., Makielski, J. C. et al. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA 286, 2264–2269 (2001).
Wilcox, R. L., Nelson, C. C., Stenzel, P. & Steiner, R. D. Postmortem screening for fatty acid oxidation disorders by analysis of Guthrie cards with tandem mass spectrometry in sudden unexpected death in infancy. J. Pediatr. 141, 833–836 (2002).
Otagiri, T., Kijima, K., Osawa, M., Ishii, K., Makita, N., Matoba, R. et al. Cardiac ion channel gene mutations in sudden infant death syndrome. Pediatr. Res. 64, 482–487 (2008).
Millat, G., Kugener, B., Chevalier, P., Chahine, M., Huang, H., Malicier, D. et al. Contribution of long-QT syndrome genetic variants in sudden infant death syndrome. Pediatr. Cardiol. 30, 502–509 (2009).
Millington, D. S., Kodo, N., Norwood, D. L. & Roe, C. R. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J. Inherit. Metab. Dis. 13, 321–324 (1990).
Shigematsu, Y., Hirano, S., Hata, I., Tanaka, Y., Sudo, M., Sakura, N. et al. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan. J. Chromatogr. B. Analyt. Technol. Biomed. Life Sci. 776, 39–48 (2002).
Yoshinaga, M., Ushinohama, H., Sato, S., Tauchi, N., Horigome, H., Takahashi, H. et al. Electrocardiographic screening of 1-month-old infants for identifying prolonged QT intervals. Circ. Arrhythm. Electrophysiol. 6, 932–938 (2013).
Yamamoto, T., Tanaka, H., Kobayashi, H., Okamura, K., Tanaka, T., Emoto, Y. et al. Retrospective review of Japanese sudden unexpected death in infancy: the importance of metabolic autopsy and expanded newborn screening. Mol. Genet. Metab. 102, 399–406 (2011).
Yamamoto, T., Mishima, H., Mizukami, H., Fukahori, Y., Umehara, T., Murase, T. et al. Metabolic autopsy with next generation sequencing in sudden unexpected death in infancy: postmortem diagnosis of fatty acid oxidation disorders. Mol. Genet. Metab. Rep. 5, 26–32 (2015).
Harrow, J., Frankish, A., Gonzalez, J. M., Tapanari, E., Diekhans, M., Kokocinski, F. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Tyner, C., Barber, G. P., Casper, J., Clawson, H., Diekhans, M., Eisenhart, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 45, D626–D634 (2017).
Mishima, H., Aerts, J., Katayama, T., Bonnal, R. J. & Yoshiura, K. The Ruby UCSC API: accessing the UCSC genome database using Ruby. BMC Bioinformatics 13, 240 (2012).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Sanguinetti, M. C. HERG1 channelopathies. Pflugers Arch. 460, 265–276 (2010).
Tan, H. L., Bardai, A., Shimizu, W., Moss, A. J., Schulze-Bahr, E., Noda, T. et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome: possible therapy implications. Circulation 114, 2096–2103 (2006).
Novotny, T., Kadlecova, J., Raudenska, M., Bittnerova, A., Andrsova, I., Florianova, A. et al. Mutation analysis ion channel genes ventricular fibrillation survivors with coronary artery disease. Pacing Clin. Electrophysiol. 34, 742–749 (2011).
Kapa, S., Tester, D. J., Salisbury, B. A., Harris-Kerr, C., Pungliya, M. S., Alders, M. et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 120, 1752–1760 (2009).
Andreasen, C., Refsgaard, L., Nielsen, J. B., Sajadieh, A., Winkel, B. G., Tfelt-Hansen, J. et al. Mutations in genes encoding cardiac ion channels previously associated with sudden infant death syndrome (SIDS) are present with high frequency in new exome data. Can. J. Cardiol. 29, 1104–1109 (2013).
Mechakra, A., Vincent, Y., Chevalier, P., Millat, G., Ficker, E., Jastrzebski, M. et al. The variant hERG/R148W associated with LQTS is a mutation that reduces current density on co-expression with the WT. Gene 536, 348–356 (2014).
Priori, S. G. & Napolitano, C. Meandering pathway leading from genotyping to personalized management of long-QT syndrome. Circulation 125, 1961–1963 (2012).
Crotti, L., Tester, D. J., White, W. M., Bartos, D. C., Insolia, R., Besana, A. et al. Long QT syndrome-associated mutations in intrauterine fetal death. JAMA 309, 1473–1482 (2013).
Nishio, Y., Makiyama, T., Itoh, H., Sakaguchi, T., Ohno, S., Gong, Y. Z. et al. D85N, a KCNE1 polymorphism, is a disease-causing gene variant in long QT syndrome. J. Am. Coll. Cardiol. 54, 812–819 (2009).
Westenskow, P., Splawski, I., Timothy, K. W., Keating, M. T. & Sanguinetti, M. C. Compound mutations: a common cause of severe long-QT syndrome. Circulation 109, 1834–1841 (2004).
Posch, M. G., Posch, M. J., Perrot, A., Dietz, R. & Ozcelik, C. Variations in DSG2: V56M, V158G and V920G are not pathogenic for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat. Clin. Pract. Cardiovasc. Med. 5, E1 (2008).
Syrris, P., Ward, D., Asimaki, A., Evans, A., Sen-Chowdhry, S., Hughes, S. E. et al. Desmoglein-2 mutations in arrhythmogenic right ventricular cardiomyopathy: a genotype–phenotype characterization of familial disease. Eur. Heart J. 28, 581–588 (2007).
Hu, D., Barajas-Martinez, H., Pfeiffer, R., Dezi, F., Pfeiffer, J., Buch, T. et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 64, 66–79 (2014).
Behr, E. R., Savio-Galimberti, E., Barc, J., Holst, A. G., Petropoulou, E., Prins, B. P. et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc. Res. 106, 520–529 (2015).
Zhang, L., Zhou, F., Huang, L., Wu, Q., Zheng, J., Wu, Y. et al. Association of common and rare variants of SCN10A gene with sudden unexplained nocturnal death syndrome in Chinese Han population. Int. J. Legal Med. 131, 53–60 (2017).
Ishikawa, T., Takahashi, N., Ohno, S., Sakurada, H., Nakamura, K., On, Y. K. et al. Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ. J. 77, 959–967 (2013).
Nakajima, T., Wu, J., Kaneko, Y., Ashihara, T., Ohno, S., Irie, T. et al. KCNE3 T4A as the genetic basis of Brugada-pattern electrocardiogram. Circ. J. 76, 2763–2772 (2012).
Kruse, M., Schulze-Bahr, E., Corfield, V., Beckmann, A., Stallmeyer, B., Kurtbay, G. et al. Impaired endocytosis of the ion channel TRPM4 is associated with human progressive familial heart block type I. J. Clin. Invest. 119, 2737–2744 (2009).
Liu, H., El Zein, L., Kruse, M., Guinamard, R., Beckmann, A., Bozio, A. et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 3, 374–385 (2010).
Daumy, X., Amarouch, M. Y., Lindenbaum, P., Bonnaud, S., Charpentier, E., Bianchi, B. et al. Targeted resequencing identifies TRPM4 as a major gene predisposing to progressive familial heart block type I. Int. J. Cardiol. 207, 349–358 (2016).
Hof, T., Liu, H., Salle, L., Schott, J. J., Ducreux, C., Millat, G. et al. TRPM4 non-selective cation channel variants in long QT syndrome. BMC Med. Genet. 18, 31 (2017).
Olson, T. M., Alekseev, A. E., Liu, X. K., Park, S., Zingman, L. V., Bienengraeber, M. et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet 15, 2185–2191 (2006).
Caldovic, L., Morizono, H. & Tuchman, M. Mutations and polymorphisms in the human N-acetylglutamate synthase (NAGS) gene. Hum. Mutat. 28, 754–759 (2007).
Al Kaabi, E. H. & El-Hattab, A. W. N-acetylglutamate synthase deficiency: novel mutation associated with neonatal presentation and literature review of molecular and phenotypic spectra. Mol. Genet. Metab. Rep. 8, 94–98 (2016).
Ohashi, Y., Hasegawa, Y., Murayama, K., Ogawa, M., Hasegawa, T., Kawai, M. et al. A new diagnostic test for VLCAD deficiency using immunohistochemistry. Neurology 62, 2209–2213 (2004).
Pie, J., Lopez-Vinas, E., Puisac, B., Menao, S., Pie, A., Casale, C. et al. Molecular genetics of HMG-CoA lyase deficiency. Mol. Genet. Metab. 92, 198–209 (2007).
Casals, N., Gomez-Puertas, P., Pie, J., Mir, C., Roca, R., Puisac, B. et al. Structural (βα)8 TIM barrel model of 3-hydroxy-3-methylglutaryl-coenzyme A lyase. J. Biol. Chem. 278, 29016–29023 (2003).
Cardoso, M. L., Rodrigues, M. R., Leao, E., Martins, E., Diogo, L., Rodrigues, E. et al. The E37X is a common HMGCL mutation in Portuguese patients with 3-hydroxy-3-methylglutaric CoA lyase deficiency. Mol. Genet. Metab. 82, 334–338 (2004).
Mir, C., Lopez-Vinas, E., Aledo, R., Puisac, B., Rizzo, C., Dionisi-Vici, C. et al. A single-residue mutation, G203E, causes 3-hydroxy-3-methylglutaric aciduria by occluding the substrate channel in the 3D structural model of HMG-CoA lyase. J. Inherit. Metab. Dis. 29, 64–70 (2006).
Yamamoto, S., Abe, H., Kohgo, T., Ogawa, A., Ohtake, A., Hayashibe, H. et al. Two novel gene mutations (Glu174—>Lys, Phe383—>Tyr) causing the ‘hepatic’ form of carnitine palmitoyltransferase II deficiency. Hum. Genet. 98, 116–118 (1996).
Yasuno, T., Kaneoka, H., Tokuyasu, T., Aoki, J., Yoshida, S., Takayanagi, M. et al. Mutations of carnitine palmitoyltransferase II (CPT II) in Japanese patients with CPT II deficiency. Clin. Genet. 73, 496–501 (2008).
Smeets, R. J., Smeitink, J. A., Semmekrot, B. A., Scholte, H. R., Wanders, R. J. & van den Heuvel, L. P. A novel splice site mutation in neonatal carnitine palmitoyltransferase II deficiency. J. Hum. Genet. 48, 8–13 (2003).
Deschauer, M., Chrzanowska-Lightowlers, Z. M., Biekmann, E., Pourfarzam, M., Taylor, R. W., Turnbull, D. M. et al. A splice junction mutation in muscle carnitine palmitoyltransferase II deficiency. Mol. Genet. Metab. 79, 124–128 (2003).
Guthrie, R. & Susi, A. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics 32, 338–343 (1963).
Millington, D. S., Kodo, N., Terada, N., Roe, D. & Chace, D. H. The analysis of diagnostic markers of genetic disorders in human blood and urine using tandem mass spectrometry with liquid secondary ion mass spectrometry. Int. J. Mass Spectrom. 111, 211–228 (1991).
Chace, D. H., Millington, D. S., Terada, N., Kahler, S. G., Roe, C. R. & Hofman, L. F. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin. Chem. 39, 66–71 (1993).
Rashed, M. S., Ozand, P. T., Harrison, M. E., Watkins, P. J. F. & Evans, S. Electrospray tandem mass-spectrometry in the diagnosis of organic acidemias. Rapid Commun. Mass Spectrom. 8, 129–133 (1994).
Rashed, M. S., Ozand, P. T., Bucknall, M. P. & Little, D. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr. Res. 38, 324–331 (1995).
Chace, D. H., Hillman, S. L., Millington, D. S., Kahler, S. G., Roe, C. R. & Naylor, E. W. Rapid diagnosis of maple syrup urine disease in blood spots from newborns by tandem mass spectrometry. Clin. Chem. 41, 62–68 (1995).
Chace, D. H., Hillman, S. L., Millington, D. S., Kahler, S. G., Adam, B. W. & Levy, H. L. Rapid diagnosis of homocystinuria and other hypermethioninemias from newborns' blood spots by tandem mass spectrometry. Clin. Chem. 42, 349–355 (1996).
Chace, D. H., Hillman, S. L., Van Hove, J. L. & Naylor, E. W. Rapid diagnosis of MCAD deficiency: quantitative analysis of octanoylcarnitine and other acylcarnitines in newborn blood spots by tandem mass spectrometry. Clin. Chem. 43, 2106–2113 (1997).
Rashed, M. S., Bucknall, M. P., Little, D., Awad, A., Jacob, M., Alamoudi, M. et al. Screening blood spots for inborn errors of metabolism by electrospray tandem mass spectrometry with a microplate batch process and a computer algorithm for automated flagging of abnormal profiles. Clin. Chem. 43, 1129–1141 (1997).
Schwartz, P. J., Stramba-Badiale, M., Crotti, L., Pedrazzini, M., Besana, A., Bosi, G. et al. Prevalence of the congenital long-QT syndrome. Circulation 120, 1761–1767 (2009).
Edmondson, A. C., Salant, J., Ierardi-Curto, L. A. & Ficicioglu, C. Missed newborn screening case of carnitine palmitoyltransferase-II deficiency. JIMD Rep. 33, 93–97 (2017).
Loporcaro, C. G., Tester, D. J., Maleszewski, J. J., Kruisselbrink, T. & Ackerman, M. J. Confirmation of cause and manner of death via a comprehensive cardiac autopsy including whole exome next-generation sequencing. Arch. Pathol. Lab. Med. 138, 1083–1089 (2014).
Bagnall, R. D., Das, K. J., Duflou, J. & Semsarian, C. Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm. 11, 655–662 (2014).
Narula, N., Tester, D. J., Paulmichl, A., Maleszewski, J. J. & Ackerman, M. J. Post-mortem whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr. Cardiol. 36, 768–778 (2015).
Hata, Y., Kinoshita, K., Mizumaki, K., Yamaguchi, Y., Hirono, K., Ichida, F. et al. Postmortem genetic analysis of sudden unexplained death syndrome under 50 years of age: a next-generation sequencing study. Heart Rhythm. 13, 1544–1551 (2016).
Probst, V., Wilde, A. A., Barc, J., Sacher, F., Babuty, D., Mabo, P. et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ. Cardiovasc. Genet 2, 552–557 (2009).
Priori, S. G., Wilde, A. A., Horie, M., Cho, Y., Behr, E. R., Berul, C. et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 10, 1932–1963 (2013).
Nielsen, M. W., Holst, A. G., Olesen, S. P. & Olesen, M. S. The genetic component of Brugada syndrome. Front. Physiol 4, 179 (2013).
Tester, D. J. & Ackerman, M. J. Genetics of long QT syndrome. Methodist. Debakey. Cardiovasc. J. 10, 29–33 (2014).
Rudic, B., Schimpf, R. & Borggrefe, M. Short QT syndrome—review of diagnosis and treatment. Arrhythm. Electrophysiol. Rev. 3, 76–79 (2014).
Schott, J. J., Alshinawi, C., Kyndt, F., Probst, V., Hoorntje, T. M., Hulsbeek, M. et al. Cardiac conduction defects associate with mutations in SCN5A. Nat. Genet. 23, 20–21 (1999).
Helbling-Leclerc, A., Bonne, G. & Schwartz, K. Emery–Dreifuss muscular dystrophy. Eur. J. Hum. Genet. 10, 157–161 (2002).
Watanabe, H., Koopmann, T. T., Le, Scouarnec, S., Yang, T., Ingram, C. R., Schott, J. J. et al. Sodium channel beta1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Invest. 118, 2260–2268 (2008).
Liu, H., El, Zein, L., Kruse, M., Guinamard, R., Beckmann, A., Bozio, A. et al. Gain-of-function mutations in TRPM4 cause autosomal dominant isolated cardiac conduction disease. Circ. Cardiovasc. Genet. 3, 374–385 (2010).
Makita, N., Seki, A., Sumitomo, N., Chkourko, H., Fukuhara, S., Watanabe, H. et al. A connexin40 mutation associated with a malignant variant of progressive familial heart block type I. Circ. Arrhythm. Electrophysiol. 5, 163–172 (2012).
Lieve, K. V., van, der Werf, C. & Wilde, A. A. Catecholaminergic polymorphic ventricular tachycardia. Circ. J. 80, 1285–1291 (2016).
Haugaa, K. H., Haland, T. F., Leren, I. S., Saberniak, J. & Edvardsen, T. Arrhythmogenic right ventricular cardiomyopathy, clinical manifestations, and diagnosis. Europace 18, 965–972 (2016).
Marcus, F. I., Edson, S. & Towbin, J. A. Genetics of arrhythmogenic right ventricular cardiomyopathy: a practical guide for physicians. J. Am. Coll. Cardiol. 61, 1945–1948 (2013).
Villoria, J. G., Pajares, S., Lopez, R. M., Marin, J. L. & Ribes, A. Neonatal screening for inherited metabolic diseases in 2016. Semin. Pediatr. Neurol. 23, 257–272 (2016).
Acknowledgements
This work was supported in part by a grant from The Ministry of Health, Labour and Welfare of Japan (26040101 to TY), grants from The Ministry of Education, Culture, Sports, Science and Technology of Japan (15H05663 to TY) and The Japan Agency for Medical Research and Development (AMED; 15km0305015h0101 to NM). We thank Ms Chisako Tsujita and Mr Takumi Osaki for their technical assistances.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Oshima, Y., Yamamoto, T., Ishikawa, T. et al. Postmortem genetic analysis of sudden unexpected death in infancy: neonatal genetic screening may enable the prevention of sudden infant death. J Hum Genet 62, 989–995 (2017). https://doi.org/10.1038/jhg.2017.79
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.79
This article is cited by
-
I536T variant of RBM20 affects splicing of cardiac structural proteins that are causative for developing dilated cardiomyopathy
Journal of Molecular Medicine (2022)
-
Accurate interpretation of genetic variants in sudden unexpected death in infancy by trio-targeted gene-sequencing panel analysis
Scientific Reports (2021)
-
Reappraisal of variants previously linked with sudden infant death syndrome: results from three population-based cohorts
European Journal of Human Genetics (2019)
-
Peri-mortem evaluation of infants who die without a diagnosis: focus on advances in genomic technology
Journal of Perinatology (2018)
