
Abstract
KBG syndrome (KBGS) is an autosomal dominant multiple congenital anomaly-intellectual disability syndrome, characterized by developmental delay with neurological involvements, macrodontia of the upper central incisors, characteristic facial dysmorphism and skeletal anomalies. Variants in ANKRD11 cause KBGS. We present five individuals from four families with ANKRD11 variants identified by whole-exome sequencing. Four of the five were clinically affected, and their diagnoses were varied. One was typical KBGS, two were Coffin–Siris syndrome-like (CSS), and one was intellectual disability with infantile spasms. One individual showed extremely mild phenotype. All individuals fulfilled the proposed diagnostic criteria for KBGS. Phenotypic features overlap between KBGS and CSS to some extent, and characteristic dental and fifth finger/toe findings can indicate differential diagnosis. These findings indicate that patients with ANKRD11 variants occupy a wide spectrum of intellectual disability, including clinically normal individuals. This is the first report highlighting the clinical overlap between KBGS and CSS and supporting the recently proposed clinical concept, in which transcriptional machineries are disrupted.
Similar content being viewed by others


Introduction
Multiple congenital anomaly-intellectual disability syndrome (MCA/ID) is a heterogeneous group of genetic syndromes comprising intellectual disability and multiple anomalies. It can be difficult to determine specific MCA/ID diagnoses for patients because of the phenotypic overlap between syndromes. Whole-exome sequencing (WES) has been successfully applied to patients with these syndromes to help diagnosis and to better understand their genetic bases.
KBG syndrome (KBGS) and Coffin–Siris syndrome (CSS) are MCA/ID syndromes that show autosomal dominant inheritance. KBGS is characterized by developmental delay with various neurological involvements, macrodontia of the upper central incisors, characteristic facial dysmorphism and skeletal anomalies.1 ANKRD11, which encodes a chromatin regulator,2 is associated with this syndrome.3 CSS is characterized by developmental delay, coarse facial appearance, feeding difficulties, recurrent infections, hypertrichosis, and hypoplastic-to-absent fifth fingernails and fifth distal phalanges.4 Genes encoding the subunits of the BAF (Brg1-associated factors) complex,5, 6 or in SOX117 are associated with CSS.
Here we present five individuals from four families with ANKRD11 variants. Four of the five are clearly affected, but have varied diagnoses: typical KBGS, CSS-like or intellectual disability with infantile spasms. We discuss their clinical phenotypes with respect to the idea of clinically distinct diseases sharing common disease mechanism disrupting transcriptional machineries.8
Materials and methods
In each family, we picked only the index individual for WES analysis as previously described.9 In brief, genomic DNA was captured using the SureSelect Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a Illumina HiSeq2000 or HiSeq2500 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Image analysis and base calling were performed using sequence control software with real-time analysis and CASAVA software (Illumina). Reads were aligned to GRCh37 using Novoalign (http://www.novocraft.com/). Marking PCR duplicates, indel realignment and base-quality-score recalibration were performed using Picard (http://picard.sourceforge.net/) and Genome Analysis ToolKit (GATK) (https://www.broadinstitute.org/gatk/index.php). Variants were called by the GATK UnifiedGenotyper (http://www.broadinstitute.org/gatk/) and annotated using ANNOVAR (http://www.openbioinformatics.org/annovar/) after excluding the common variants registered in the common dbSNP135 database (minor allele frequency ⩾0.01). Detected AKNRD11 variants were validated by Sanger sequencing. Parental samples were also tested to check the mode of inheritance of identified variants. Experimental protocols were approved by the Committee for Ethical Issues at Yokohama City University School of Medicine. Written informed consent was obtained from all individuals or their parents. Clinical information was collected from medical records obtained by doctor–patient consultation.
Results
Genetic analysis
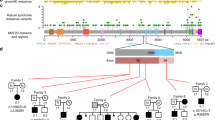
WES with the depth of 10 × reads or more covered 90.5–93.5% of the total coding sequence of RefSeq genes. Through WES, we identified three novel variants in ANKRD11 (NM_013275.4): a de novo 4 bp deletion (c.3224_3227del: p.Glu1075Glyfs*242) in individual 2, a de novo nonsense variant (c.6187G>T: p.Glu2063*) in individual 3, and a missense variant (c.6416C>T: p.Pro2139Leu) in individual 4 that was also present in his mother (individual 5). We also identified a reported variant10 (c.1903_1907del: p.Lys635Glnfs*26) in individual 1 that had arisen de novo (Figure 1). Pro2139 is a well-conserved amino acid among vertebrate species and it resides in the activation domain, which is capable of stimulating transcription.11 Web-based tools (SIFT, PolyPhen2 and MutationTaster) predicted that this variant was pathogenic (SIFT: 0, PolyPhen2 (HumVar): probably damaging with score 0.999, and MutationTaster: disease causing with a probability >0.999). This variant was not detected in 575 normal Japanese control exomes, or in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), the 1000 Genome Project (http://www.1000genomes.org/), or the Human Genetics Variation Database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/). In ExAC browser (http://exac.broadinstitute.org/), only 2 out of 106 544 alleles (minor allele frequency=1.87 × 10−5) were detected to have this variant in a heterozygous state. Considering that individual 5 had an extremely mild KBGS phenotype, the pathogenicity of this variant in this family could not be excluded. In individuals 1 and 2 with the initial diagnosis of CSS-like, no variants were identified in any of BAF complex-related gene or in SOX11. In individual 4 who was initially suspected to have Noonan syndrome, no variants were identified in genes associated with Noonan syndrome (PTPN11, SOS1, RAF1, RIT1, KRAS, NRAS, BRAF, MAP2K1, RRAS, RASA2, A2ML1, SOS2andLZTR1).

ANKRD11 functional domains and detected variants. Upper section depicts ANKRD11 protein structure. AD, activation domain; ANK, ankyrin repeats; RD1, repression domain 1; RD2, repression domain 2. Middle section shows the location of identified variants. Lower section shows electropherograms and substituted/truncated amino acids found in each individual. ANKRD11 sequence is based on RefSeq NM_013275.4. Note that Sanger sequencing of the PCR amplicons at the mutation site in individual 5 showed the same height of mutant and wild-type allele peaks, respectively, implying the possible heterozygote. A full color version of this figure is available at the Journal of Human Genetics journal online.
Phenotypic analysis
The clinical features of the five individuals are presented in Figure 2,Supplementary Figure 2, Table 1 and Supplementary Table 1. Individuals 4 and 5 did not give us permission to present their photographs. Individuals 1 and 2 were clinically diagnosed as CSS-like. Individual 3 was not specifically diagnosed, and was considered as a patient with ID and infantile spasms of an unknown cause. Individual 4 was clinically diagnosed as KBGS. Individual 5 was suspected to have mild KBGS after finding the variant. She has never had developmental delay nor specific medical history, and currently has normal intelligence, but she has short stature, facial dysmorphism and macrodontia of upper central incisors with the width of 12–13 mm.

Clinical features of individuals with KBG syndrome. (a) Images of individual 1 at 7 years of age. From left to right: frontal view, lateral view, right foot and left hand. Hypertelorism and prominent lips are present. Brachydactyly of the first finger and toe is recognized, as well as sandal gap in the foot. Fifth finger/toe hypoplasia and nail hypoplasia are not seen. (b) Images of individual 2 at age 10 years. From left to right: frontal view with his mouth open, frontal view with his mouth closed, lateral view, occipital view and left hand. Thick eyebrows, long eyelashes, inverted epicanthal folds, broad nasal bridge with bulbous nasal tip, thick lips and low posterior hairline were observed. Brachydactyly of all fingers and clinodactyly of the fifth finger were present. (c) Images of individual 3 at age 1 or 2 years. From left to right: frontal view at 1-year old with her mouth closed, frontal view at age 2 years with her mouth slightly open, lateral view at age 2 years, and T1-weighted sagittal brain magnetic resonance image (MRI) performed at age 2 years. Brachycephaly with a round facial shape, narrow palpebral fissures, prominent nasal bridge, smooth philtrum and down turned corners of month are present. Brain MRI shows a markedly intense, curvilinear lesion stretching over the superior margin of the corpus callosum body through the posterior margin of the splenium to the retro-thalamic cistern, which was diagnosed as pericallosal lipoma, and cerebellar vermis hypoplasia. (d) Dental findings of individuals 1–4. From upper left to right: individual 1 at age 7 years, individual 2 at age 10 years and individual 3 at age 2 years. In individual 1, primary teeth are of normal shape and size. In individual 2 macrodontia of upper incisors, supramentary, malpositioned mamelons are present. In individual 3 upper deciduous incisors are large. From lower left to right: individual 4 at age 7 years 1 month, and his pan tomography at that time. In individual 4 upper incisors are large and supramentary mamelons are noted. A full color version of this figure is available at the Journal of Human Genetics journal online.
Among the five individuals, frequently observed characteristics were delayed bone age (3/3; 100%), macrodontia (4/5; 80%), speech delay (3/5; 60%), short stature (3/5; 60%), hand anomalies (3/5; 60%), neurodevelopmental delay (3/5; 60%), seizures (3/5; 60%), hearing loss (3/5; 60%) and vision anomalies (3/5; 60%). Common facial dysmorphic features were round/triangular face (4/5; 80%), hairline abnormality (4/5; 80%), hypertelorism (4/5; 80%), synophrys (3/5; 60%) and prominent nasal bridge (3/5; 60%).
Macrodontia was the most frequently observed dental abnormality, and supramentary, malpositioned mamelons were observed in individuals 2 and 4. Individual 1 had abnormally delayed dentition, which is a characteristic feature of CSS. Two of the five individuals had costovertebral abnormality (abnormal vertebrae and scoliosis in individual 4, and spinal abnormality in individual 3). Hand anomalies in the five individuals were short hand and tubular bones, and brachydactyly in three of four, and two of three individuals, respectively. Brachydactyly–clinodactyly of the fifth finger without hypoplasticity was noted in individuals 1 and 2, who were CSS-like. Severity in neurodevelopmental delay varied among the five individuals. Cognitive delay was observed in three of the five individuals, together with speech delay. Speech delay was relatively severe. Individual 1 could use several words at age 7 years; individual 2 attained sentence-level speech at age 10 years; and individual 3 spoke no words at 2 years of age. These three also had seizures. Individuals 4 and 5 were evaluated to be normal with respect to neurodevelopment. With regard to behavioral abnormalities, individual 2 had pervasive developmental disorders and individual 3 was suspected to have autism spectrum disorder, but she was too young to be diagnosed as autism spectrum disorder.
In CSS-like individuals, several CSS features, such as ‘coarse face’, thick lower lip, wide mouth, long eyelashes, ear abnormality and abnormal delayed dentition, were observed, while feeding difficulty, hypotonia, sparse hair, and absent/hypoplastic fifth finger/nail, joint laxity, body hypertrichosis, and recurrent infections were not observed.
Pericallosal lipoma was identified in individual 3 by brain MRI. This has not been previously described in KBGS.
All five individuals with an ANKRD11 variant fulfilled the established diagnostic criteria for KBGS (four or more of eight major criteria).12 The recently proposed, modified criteria10 were also satisfied in all five individuals.
Discussion
We reviewed the literature and compared the representative characteristics of molecularly confirmed patients with these two syndromes (Table 1, Supplementary Table 1 and Supplementary Figure 1).3, 10, 13, 14, 15, 16 The phenotypic features of CSS overlap those of KBGS to some extent. These include growth abnormality (short stature, delayed bone age), neurodevelopmental delay, behavioral abnormalities, seizures, hearing loss or brain MRI abnormality (Supplementary Figure 1a). However we have to be aware that these overlapping features are quite unspecific and would be observed in most MCA/IDs. Both KBGS and CSS can show facial dysmorphism, dental and fifth finger/toe malformation, but their expression seems to be different with a minimal overlap (Supplementary Figure 1b and c). Patients with KBGS tend to have facial characteristics of brachycephaly, round/triangular face, bulbous nasal tip, prominent nasal bridge, hypertelorism and synophrys. On the other hand, sparse hair, thick eyebrows, long eyelashes, wide mouth and thick lower lip, which are all ‘coarse face’ features, are more frequently observed in CSS (Supplementary Figure 1c). Other features of KGBS that are useful for differential diagnosis are macrodontia, brachy/clinodactyly of fifth finger/toe, short tubular bones of the hand, and first-degree relatives of KBGS probands who are also affected. Meanwhile, CSS characteristics that often distinguish from KBGS include delayed dentition, absent/hypoplastic fifth finger/toe/nail, body hypertrichosis, feeding difficulty, hypotonia and recurrent infection (Supplementary Figure 1b). Patients 1 and 2 were clinically diagnosed as CSS-like but had mixed phenotypes of KBGS and CSS. It was noted that delayed eruption of primary dentition were observed in individual 1 as a new dental finding for KBGS. Previously individual 4 was clinically suspected to have Noonan syndrome based on the findings of short stature, hypertelorism, webbed short neck and cryptorchidism. He also had congenital heart defects including aortic stenosis, mitral regurgitation and mitral stenosis, which seemed atypical (retrospectively) as Noonan syndrome in a sense that they were all left-side defects in heart.
Intrafamilial variability of KBGS has been reported,17 and more male cases have been reported than females in a family probably due to the different expressivity. Individuals 4 and 5 are another examples. Further clinical investigation revealed that the mother of individual 5 also had short stature (148 cm: −2.0 s.d., calculating using the data from National Health and Nutrition Survey in Japan, 2014, on the mean height (158.3 cm) and s.d. (5.1) for females with ages 30–39 years), and she resembles her daughter with very mild dysmorphic face. Unfortunately we could not obtain DNA from parents of individual 5 for further investigation. Considering individual 4 as a typical KBGS phenotype and intrafamilial variability in KBGS, the missense variant found in this family is probably pathogenic. The variant is supported as pathogenic by the following evidences: the extreme rarity in general population, in silico prediction as pathogenic and occurrence at conserved amino acid within the domain. There has been two reported patients with KBGS having a missense variant in AKNRD11.16, 18 They satisfied the diagnostic criteria (Supplementary Table 2). We compared the phenotypes of the four individuals (two from the literature, and individuals 4 and 5 from our study) with KBGS having a missense ANKRD11 variant, to those with truncating ANKRD11 variants (Supplementary Table 3). The former tended to express typical phenotypes, except the cognitive delay, observed in relatively low frequency (25% in missense group vs 85.7% in truncation group). Further studies of KBGS patients with missense variants are needed to clarify this issue.
Another possible explanation for the mild phenotype observed in individual 5 is the somatic mosaicism. However, Sanger sequencing of the PCR amplicons at the mutation site showed the same height of mutant and wild-type peaks amplified from mutant and wild-type alleles, respectively, implying the possible heterozygote rather than the somatic mosaic mutation. DNA samples from different tissues of individual 5 were unavailable for further investigation.
Three of the individuals had mild to moderate motor/cognitive deficits and relatively severe speech difficulties. On the other hand, individuals 4 and 5 had no neurodevelopmental abnormalities. According to our literature review, 32.3% of patients with KBGS had affected first-degree relatives. Considering the highly varied clinical severity of KBGS, with the mildest form, KBGS can be inherited. In contrast, CSS is essentially sporadic and is rarely inherited.
Genetic analysis of syndromic intellectual disabilities using next-generation sequencing has identified overlaps among some MCA/ID syndromes where mutant genes are associated with epigenetic modification; for example between CSS and Nicolaides–Baraitser syndrome sharing SMARCA2 variant,19 CSS and Borjeson–Forssman–Lehmann syndrome with PHF6 variant,20 Kabuki syndrome, Wiedemann–Steiner syndrome (WSS) and Cornelia de Lange syndrome (CdLS) with KMT2A variant,8, 21 and Cornelia de Lange syndrome (CdLS) and KBGS with ANKRD11 variant.15, 22 Our study suggests that it might be sometimes difficult to clinically distinguish between CCS and KBGS. In mice, ANKRD11 is a chromatin regulator that binds to histone deacetylase 3 (HDAC3) and controls histone acetylation and the expression of multiple genes that are important for neural development.2 The BAF complex is an ATP-dependent chromatin remodeling complex that is essential for neural crest development.23 This is the first report presenting phenotypic overlap between KBGS and CSS, adding more evidence for the possibility of two distinct diseases sharing a common genetic mechanism of disrupting transcriptional machineries.
In conclusion, variants in ANKRD11 cause a wide spectrum of clinical phenotypes, even with intrafamilial phenotypic variability, from the mild form of KBGS to CSS-like syndrome.
Ethics statement
We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
References
Herrmann, J., Pallister, P. D., Tiddy, W. & Opitz, J. M. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects Orig. Artic. Ser. 11, 7–18 (1975).
Gallagher, D., Voronova, A., Zander, M. A., Cancino, G. I., Bramall, A., Krause, M. P. et al. Ankrd11 is a chromatin regulator involved in autism that is essential for neural development. Dev. Cell 32, 31–42 (2015).
Sirmaci, A., Spiliopoulos, M., Brancati, F., Powell, E., Duman, D., Abrams, A. et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 89, 289–294 (2011).
Coffin, G. S. & Siris, E. Mental retardation with absent fifth fingernail and terminal phalanx. Am. J. Dis. Child. 119, 433–439 (1970).
Santen, G. W., Aten, E., Sun, Y., Almomani, R., Gilissen, C., Nielsen, M. et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380 (2012).
Tsurusaki, Y., Okamoto, N., Ohashi, H., Kosho, T., Imai, Y., Hibi-Ko, Y. et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–378 (2012).
Tsurusaki, Y., Koshimizu, E., Ohashi, H., Phadke, S., Kou, I., Shiina, M. et al. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat. Commun. 5, 4011 (2014).
Yuan, B., Pehlivan, D., Karaca, E., Patel, N., Charng, W. L., Gambin, T. et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J. Clin. Invest. 125, 636–651 (2015).
Yoshida, K., Miyatake, S., Kinoshita, T., Doi, H., Tsurusaki, Y., Miyake, N. et al. 'Cortical cerebellar atrophy' dwindles away in the era of next-generation sequencing. J. Hum. Genet. 59, 589–590 (2014).
Ockeloen, C. W., Willemsen, M. H., de Munnik, S., van Bon, B. W., de Leeuw, N., Verrips, A. et al. Further delineation of the KBG syndrome phenotype caused by ANKRD11 aberrations. Eur. J. Hum. Genet. 23, 1178–1185 (2014).
Zhang, A., Li, C. W. & Chen, J. D. Characterization of transcriptional regulatory domains of ankyrin repeat cofactor-1. Biochem. Biophys. Res. Commun. 358, 1034–1040 (2007).
Skjei, K. L., Martin, M. M. & Slavotinek, A. M. KBG syndrome: report of twins, neurological characteristics, and delineation of diagnostic criteria. Am. J. Med. Genet. A 143, 292–300 (2007).
Kim, H. J., Cho, E., Park, J. B. & Im, W. Y. A Korean family with KBG syndrome identified by ANKRD11 mutation, and phenotypic comparison of ANKRD11 mutation and 16q24.3 microdeletion. Eur. J. Med. Genet. 58, 86–94 (2015).
Samanta, D. & Willis, E. Electroencephalographic findings in KBG syndrome: a child with novel mutation in ANKRD11 gene. Acta Neurol. Belg. 115, 779–782 (2015).
Parenti, I., Gervasini, C., Pozojevic, J., Graul-Neumann, L., Azzollini, J., Braunholz, D. et al. Broadening of cohesinopathies: exome sequencing identifies mutations in ANKRD11 in two patients with Cornelia de Lange-overlapping phenotype. Clin. Genet. 89, 74–81 (2016).
Walz, K., Cohen, D., Neilsen, P. M., Foster, J. 2nd, Brancati, F., Demir, K. et al. Characterization of ANKRD11 mutations in humans and mice related to KBG syndrome. Hum. Genet. 134, 181–190 (2015).
Brancati, F., Sarkozy, A. & Dallapiccola, B. KBG syndrome. Orphanet J. Rare Dis. 1, 50 (2006).
Xu, M., Zhou, H., Yong, J., Cong, P., Li, C., Yu, Y. et al. A Chinese patient with KBG syndrome and a 9q31.2-33.1 microdeletion. Eur. J. Med. Genet. 56, 245–250 (2013).
Wieczorek, D., Bogershausen, N., Beleggia, F., Steiner-Haldenstatt, S., Pohl, E., Li, Y. et al. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum. Mol. Genet. 22, 5121–5135 (2013).
Zweier, C., Rittinger, O., Bader, I., Berland, S., Cole, T., Degenhardt, F. et al. Females with de novo aberrations in PHF6: clinical overlap of Borjeson-Forssman-Lehmann with Coffin-Siris syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 166C, 290–301 (2014).
Miyake, N., Tsurusaki, Y., Koshimizu, E., Okamoto, N., Kosho, T., Brown, N. J. et al. Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin. Genet. 89, 115–119 (2016).
Ansari, M., Poke, G., Ferry, Q., Williamson, K., Aldridge, R., Meynert, A. M. et al. Genetic heterogeneity in Cornelia de Lange syndrome (CdLS) and CdLS-like phenotypes with observed and predicted levels of mosaicism. J. Med. Genet. 51, 659–668 (2014).
Santen, G. W., Kriek, M. & van Attikum, H. SWI/SNF complex in disorder: SWItching from malignancies to intellectual disability. Epigenetics 7, 1219–1224 (2012).
Acknowledgements
We thank all the participants for their cooperation in this research. We also thank Ms K Takabe, Mr T Miyama, Ms N Watanabe, Ms M Sato, Mr S Nakamura and Ms S Sugimoto at the Department of Human Genetics, Yokohama City University Graduate School of Medicine, for their technical assistance. This work was supported by grants from Research on Measures for Intractable Diseases (N Matsumoto), Comprehensive Research on Disability Health and Welfare (N Matsumoto), the Strategic Research Program for Brain Science (N Matsumoto), the Initiative on Rare and Undiagnosed Diseases in Pediatrics (N Matsumoto), the Initiative on Rare and Undiagnosed Diseases for Adults (N Matsumoto) from the Japanese Agency for Medical Research and Development, a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Grants-in-Aid for Scientific Research (B (N Miyake and H Saitsu) and C (S Miyatake)); the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems (N Matsumoto) from the Japanese Science and Technology Agency; the Takeda Science Foundation (N Miyake, H Saitsu and N Matsumoto); and The Ichiro Kanehara Foundation for the Promotion of Medical Science & Medical Care (S Miyatake).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Miyatake, S., Okamoto, N., Stark, Z. et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin–Siris-like syndrome. J Hum Genet 62, 741–746 (2017). https://doi.org/10.1038/jhg.2017.24
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.24
This article is cited by
-
Integrated in silico MS-based phosphoproteomics and network enrichment analysis of RASopathy proteins
Orphanet Journal of Rare Diseases (2021)
-
A de novo frameshift variant of ANKRD11 (c.1366_1367dup) in a Chinese patient with KBG syndrome
BMC Medical Genomics (2021)
-
PEX10-related autosomal recessive cerebellar ataxia with hearing loss
Acta Neurologica Belgica (2020)
-
A novel SLC9A1 mutation causes cerebellar ataxia
Journal of Human Genetics (2018)
-
Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations
Journal of Human Genetics (2018)

{kind=link}
{kind=link}