
Abstract
MEIS2 aberrations are considered to be the cause of intellectual disability, cleft palate and cardiac septal defect, as MEIS2 copy number variation is often observed with these phenotypes. To our knowledge, only one nucleotide-level change—specifically, an in-frame MEIS2 deletion—has so far been reported. Here, we report a female patient with a de novo nonsense mutation (c.611C>G, p.Ser204*) in MEIS2. She showed severe intellectual disability, moderate motor/verbal developmental delay, cleft palate, cardiac septal defect, hypermetropia, severe feeding difficulties with gastro-esophageal reflux and constipation. By reviewing this patient and previous patients with MEIS2 point mutations, we found that feeding difficulty with gastro-esophageal reflux appears to be one of the core clinical features of MEIS2 haploinsufficiency, in addition to intellectual disability, cleft palate and cardiac septal defect.
Similar content being viewed by others

Introduction
Meis homeobox 2 (MEIS2; also known as MRG1, NM_170677.4) encodes a homeobox (HOX) protein belonging to the three amino acid loop extension (TALE) superfamily. TALE homeobox proteins are highly conserved transcription factors.1 MEIS functions as a HOX co-factor that cooperatively binds deoxyribonucleic acid (DNA) to pre-B cell leukemia transcription factor (PBX) and/or HOX proteins to increase the affinity and specificity of their DNA binding activity.2, 3 In mice, Meis2 is expressed in embryonic neural crest cells (multipotent and migratory cells), particularly in the heart, central nervous system, upper and lower jaw, and intestinal tract.4 Meis2 null mice are embryonic lethal at E13.5–14.5, with defects of craniofacial cartilage and heart derived from neural crest cells, which is also observed in knockdown zebrafish.4, 5, 6 The detailed phenotype of heterozygous knockout mice is unknown. Human MEIS2 is highly expressed in fetal and adult brain and lymphoid organs including spleen, lymph node, thymus and appendix.7, 8 Further, MEIS2 mutations in humans may lead to cardiac septal defects, cleft palate and intellectual disability (ID), as observed with MEIS2 haploinsufficiency in six sporadic and one familial deletion cases (123 kb–5.6 Mb), and in another family with a 58-kb duplication that results in a frame shift mutation.9, 10, 11, 12 Only one MEIS2 mutation at the nucleotide level has been found in a female patient: c.998_1000del (p.Arg333del).13 This 3-bp deletion at the homeodomain may interfere with DNA binding. Moreover, this female patient also presented with various anomalous features and developmental abnormalities. Here, we report on another MEIS2 mutation case of specific clinical features and discuss the clinical consequence of MEIS2 mutations.
Case Report
The patient is the first child of non-consanguineous healthy French parents. She was born after a full-term uneventful pregnancy. Her birth length was 46 cm (−2.5SD), weight 3270 g (mean) and head circumference 33.5 cm (−1SD). Cleft palate, atrial septal defect (ASD) and ventricular septal defect (VSD) were noted at birth. ASD and VSD were surgically corrected 2 weeks after birth, and her cleft palate was operated on twice (at 7 and 23 months). She presented with mild morphological features including a large forehead, mild trigonocephaly, sparse eyebrow, deeply set eyes, large and low-set ears, full cheeks and thin upper lip vermilion (Figures 1a–c). She had a serious feeding difficulty with severe gastro-esophageal reflux requiring gastrostomy at 3 months. Her motor development was delayed: sitting at 11 months and independent gait at 2 years 9 months. Severe hypermetropia was also noticed. At 2 years 9 months, her height was 95 cm (+1SD), weight 12.5 kg (mean) and head circumference 46.8 cm (−1.5SD). Her speech was delayed with only a few words at 2 years 9 months and severe constipation was noted. At 18 months, brain magnetic resonance imaging and comparative genomic hybridization array (Agilent 60k, Agilent Technologies, Santa Clara, CA, USA) were all normal.

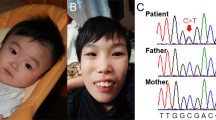
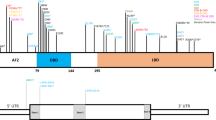
Patient’s facial features and MEIS2 mutation. Facial photographs at 13 months (a) and 20 months (b and c). (d) Electoropherograms for the c.611C>G (p.Ser204*) mutation in MEIS2. Arrow indicates the mutation. (e) Schematic representation of MEIS2 protein with its domain and the mutation. Open and filled circles indicate a previously reported mutation and the current mutation, respectively. The protein contains a N-terminal of Homeobox Meis and PKNOX1 (Meis_PKNOX_N) domain and a homeobox (HOX) domain predicted by SMART (http://smart.embl.de/). A full color version of this figure is available at the Journal of Human Genetics journal online.
Materials and methods
Whole-exome sequencing was performed for the patient only. Blood leukocytes were obtained from the patient and her parents with their informed consent. Genomic DNA was extracted by standard methods, partitioned using the SureSelect Human All Exon v5 Kit (Agilent Technologies), and sequenced by Illumina HiSeq2000 (Illumina, San Diego, CA, USA) with 101-bp paired-end reads. Variants were selected by excluding synonymous variants and variants registered in dbSNP137, our in-house exome database of 575 Japanese individuals, and the NHLBI Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/) (MAF≧0.01). The remaining variants were then evaluated depending on whether they fitted an X-linked or autosomal dominant model (de novo occurrence), and were found in the known mutant genes registered in the Human Gene Mutation database Professional (http://www.biobase-international.com/product/hgmd). Candidate variants were confirmed by Sanger sequencing. Parentage was tested using nine microsatellite markers with Gene Mapper software v4.1.1 (Life Technologies Inc., Carlsbad, CA, USA). This study was approved by the institutional review board of Yokohama City University School of Medicine.
Results and Discussion
Mean whole-exome sequencing read depth in protein coding regions of RefSeq was 102 × , with 93.9% of target regions covered by 20 reads or more. Through our variant selection, we successfully narrowed down the variants to a heterozygous nonsense MEIS2 mutation (c.611C>G, p.Ser204*). In addition, using Sanger sequencing we confirmed that the mutation occurred de novo with no discrepant parentage (Figure 1d).
Similar to patients with MEIS2 haploinsufficiency, two patients possessing MEIS2 point mutations commonly show cleft palate, cardiac septal defect and ID (Table 1).9, 10, 11, 12, 13 Therefore, we reconfirm these clinical features as the MEIS2 alteration phenotype. In addition, severe gastro-esophageal reflux requiring gastrostomy was observed commonly in both patients with a MEIS2 mutation (Table 1).13 MEIS proteins play an important role in neural crest cell development.2, 4 Neural crest cell-related diseases are collected under the term “neurocristopathy”, and include the 22q11.2 deletion syndrome, CHARGE syndrome and Hirschsprung disease, which is a congenital intestinal motility disorder.14, 15 Like MEIS2 alterations, digestive symptoms (gastro-esophageal reflux, esophagitis or chronic constipation) are observed in 24% (50/211) of patients with 22q11.2 deletions,16 and gastro-esophageal reflux is observed in 89% (32/36) of patients with CHARGE syndrome.17 Therefore, digestive symptoms may be a part of the phenotype caused by a loss-of-function mutation in MEIS2, and may result from neuronal dysfunction and/or abnormal number of neurons as seen in the neurocristopathy.15, 17 Nevertheless, to fully understand the disease accumulation of patients with MEIS2 aberrations is highly encouraged.
In summary, we have identified a novel nonsense MEIS2 mutation in a female patient with cleft palate, cardiac septal defect, severe ID, developmental delay and feeding difficulty with gastro-esophageal reflux. Our findings strongly suggest neurocristopathy be included in the clinical features associated with MEIS2 alterations.
References
Yang, Y., Hwang, C. K., D'Souza, U. M., Lee, S. H., Junn, E. & Mouradian, M. M. Three-amino acid extension loop homeodomain proteins Meis2 and TGIF differentially regulate transcription. J. Biol. Chem. 275, 20734–20741 (2000).
Geerts, D., Schilderink, N., Jorritsma, G. & Versteeg, R. The role of the MEIS homeobox genes in neuroblastoma. Cancer Lett. 197, 87–92 (2003).
Mann, R. S., Lelli, K. M. & Joshi, R. Hox specificity unique roles for cofactors and collaborators. Curr. Top. Dev. Biol. 88, 63–101 (2009).
Machon, O., Masek, J., Machonova, O., Krauss, S. & Kozmik, Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol. 15, 40 (2015).
Paige, S. L., Thomas, S., Stoick-Cooper, C. L., Wang, H., Maves, L., Sandstrom, R. et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 151, 221–232 (2012).
Melvin, V. S., Feng, W., Hernandez-Lagunas, L., Artinger, K. B. & Williams, T. A morpholino-based screen to identify novel genes involved in craniofacial morphogenesis. Dev. Dyn. 242, 817–831 (2013).
Larsen, K. B., Lutterodt, M. C., Laursen, H., Graem, N., Pakkenberg, B., Mollgard, K. et al. Spatiotemporal distribution of PAX6 and MEIS2 expression and total cell numbers in the ganglionic eminence in the early developing human forebrain. Dev. Neurosci. 32, 149–162 (2010).
Smith, J. E., Afonja, O., Yee, H. T., Inghirami, G. & Takeshita, K. Chromosomal mapping to 15q14 and expression analysis of the human MEIS2 homeobox gene. Mamm. Genome 8, 951–952 (1997).
Erdogan, F., Ullmann, R., Chen, W., Schubert, M., Adolph, S., Hultschig, C. et al. Characterization of a 5.3 Mb deletion in 15q14 by comparative genomic hybridization using a whole genome “tiling path” BAC array in a girl with heart defect, cleft palate, and developmental delay. Am. J. Med. Genet. A 143, 172–178 (2007).
Chen, C. P., Lin, S. P., Tsai, F. J., Chern, S. R., Lee, C. C. & Wang, W. A 5.6-Mb deletion in 15q14 in a boy with speech and language disorder, cleft palate, epilepsy, a ventricular septal defect, mental retardation and developmental delay. Eur. J. Med. Genet. 51, 368–372 (2008).
Crowley, M. A., Conlin, L. K., Zackai, E. H., Deardorff, M. A., Thiel, B. D. & Spinner, N. B. Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am. J. Med. Genet. A 152, 1326–1327 (2010).
Johansson, S., Berland, S., Gradek, G. A., Bongers, E., de Leeuw, N., Pfundt, R. et al. Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am. J. Med. Genet. A 164, 1622–1626 (2014).
Louw, J. J., Corveleyn, A., Jia, Y., Hens, G., Gewillig, M. & Devriendt, K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am. J. Med. Genet. A 167, 1142–1146 (2015).
Sanlaville, D. & Verloes, A. CHARGE syndrome: an update. Eur. J. Hum. Genet. 15, 389–399 (2007).
Zhang, D., Ighaniyan, S., Stathopoulos, L., Rollo, B., Landman, K., Hutson, J. et al. The neural crest: a versatile organ system. Birth Defects Res. C Embryo Today 102, 275–298 (2014).
Eicher, P. S., McDonald-Mcginn, D. M., Fox, C. A., Driscoll, D. A., Emanuel, B. S. & Zackai, E. H. Dysphagia in children with a 22q11.2 deletion: unusual pattern found on modified barium swallow. J. Pediatr. 137, 158–164 (2000).
Dobbelsteyn, C., Peacocke, S. D., Blake, K., Crist, W. & Rashid, M. Feeding difficulties in children with CHARGE syndrome: prevalence, risk factors, and prognosis. Dysphagia 23, 127–135 (2008).
Acknowledgements
We would like to thank the patient and her family for participating in this study. We also thank Ms. S Sugimoto and K Takabe for their technical assistance. This work was supported in part by a grant for Research on Measures for Intractable Diseases; a grant for Comprehensive Research on Disability Health and Welfare, the Strategic Research Program for Brain Science (SRPBS), a grant for Initiative on Rare and Undiagnosed Diseases in Pediatrics from Japan Agency for Medical Research and Development (AMED); a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); Grants-in-Aid for Scientific Research (B and C) and Challenging Exploratory Research from the Japan Society for the Promotion of Science (JSPS); the fund for Creation of Innovation Centers for Advanced Interdisciplinary Research Areas Program in the Project for Developing Innovation Systems from the Japan Science and Technology Agency (JST); the Takeda Science Foundation; the Yokohama Foundation for Advancement of Medical Science; and the Hayashi Memorial Foundation for Female Natural Scientists.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Fujita, A., Isidor, B., Piloquet, H. et al. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J Hum Genet 61, 835–838 (2016). https://doi.org/10.1038/jhg.2016.54
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.54
This article is cited by
-
MEIS2 (15q14) gene deletions in siblings with mild developmental phenotypes and bifid uvula: documentation of mosaicism in an unaffected parent
Molecular Cytogenetics (2021)
-
Duplications involving the long range HMX1 enhancer are associated with human isolated bilateral concha-type microtia
Journal of Translational Medicine (2020)
-
De novo truncating variants in PHF21A cause intellectual disability and craniofacial anomalies
European Journal of Human Genetics (2019)
-
Heterozygous loss-of-function variants of MEIS2 cause a triad of palatal defects, congenital heart defects, and intellectual disability
European Journal of Human Genetics (2019)
-
MEIS2 regulates endothelial to hematopoietic transition of human embryonic stem cells by targeting TAL1
Stem Cell Research & Therapy (2018)
