
Abstract
Although in the last two decades there has been considerable progress in understanding the genetic basis of Parkinson’s disease (PD), the majority of PD is sporadic and its genetic causes are largely unknown. In an attempt to identify novel genetic causes of PD, whole-exome sequencing and subsequent analyses were performed in a family featuring late-onset PD with cognitive impairment. A novel genetic variant (p.Arg610Gly) in the GIGYF2 gene, previously known to be associated with PD, was identified as potential disease-causing mutation. The GIGYF2 p.Arg610Gly mutation situated in the GYF domain of the encoding protein was predicted to be pathogenic and to disrupt the GYF’s ligand-binding abilities. Although further research is still required, this finding may shed light on the GIGYF2-associated mechanisms that lead to PD and suggests insulin dysregulation as a disease-specific mechanism for both PD and cognitive dysfunction.
Similar content being viewed by others

Main
Parkinson’s disease (PD) is the second most common neurodegenerative disease behind Alzheimer disease and affects more than four million people worldwide. Although in the last two decades there has been considerable progress in understanding the genetic basis of PD, its pathogenic causes are largely unknown.1 In this study, we aimed to identify novel genes causing PD by performing whole-exome sequencing and subsequent analyses in a Spanish family featuring a late-onset form of PD (Figure 1). The age at onset of our family ranged from 78 to 88 years old and the clinical phenotype was characterized by the presence of a mild motor parkinsonism with an unilateral tremor in one member, a rigid-akinetic unilateral syndrome in a second member, and a jaw tremor in a third member. Even though jaw tremor has been considered a symptom of essential tremor, when it appears in essential tremor is probably a marker for subsequent conversion to PD.2 Cognitive impairment also occurred in all affected individuals. See Supplementary Online Material for more clinical details.

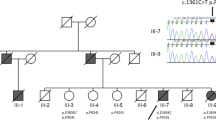
GIGYF2 mutation in LOPD. Owing to the late-onset of the disease presentation, ranging from 78 to 88 years, and the early death of apparently unaffected parents (age of death: 72 and 43 years for father and mother, respectively), the pattern of inheritance in this family remains unknown. (a) Pedigree structure of the family analyzed in this study. wt/m: heterozygous mutation carrier. WES was performed in individuals highlighted with a blue asterisk. (b) GIGYF2 p.Arg610Gly mutation: chromatogram sequences of wild-type (top) and mutant (bottom) sequences are shown on the right side, whereas conservation of the GIGYF2 p.Arg610Gly mutation in different species and GIGYF1 protein is shown on the left side. HS: Homo sapiens; CL: Canis lupus; BT: Bos taurus; MM: Mus musculus; DR: Danio rerio. (c) GIGYF2 protein structure predicted by SMART (http://smart.embl-heidelberg.de/). The GYF domain consists of highly conserved glycine–tyrosine–phenylanine residues following the ‘GP[YF]xxxx[MV]xxWxxx[GN]YF’ motif containing 60 amino acids. The R610 amino acid (highlighted in red) is the last residue of the GYF domain. A full color version of this figure is available at the Journal of Human Genetics journal online.
Whole-exome sequencing was performed in two affected siblings by using the SureSelectXT Human All exon 50 Mb exon-capture kit (Agilent Technologies Inc., Santa Clara, CA, USA) and HiSeq 2000 following the manufacturer's instructions for paired-end 150-bp reads (Illumina Inc., San Diego, CA, USA). Whole-exome sequencing data were then processed and analyzed through a computational pipeline following the general workflow adopted by the 1000 genomes project.3 Ninety-one percent of the target exome was captured at 30-fold coverage or higher in both patients. Common genetic variation (frequency >3%) observed in the latest dbSNP137 build, 1000 Genomes Project Phase 1, other public databases, such as the Exome Variant Server of the National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project,4 and exomes generated in house3 were removed from further analyses. Sanger sequencing was used for single-nucleotide variant (SNV) call validation and disease-segregation analyses. Although four novel SNVs were identified present in the three affected individuals and absent in large number of control individuals (>10 000), including 188 ethnicity-matched control chromosomes, only one SNV, located in the GIGYF2 gene and not present in the Exome Aggregation Consortium, was predicted to be pathogenic (Table 1). While the pathogenic role of GIGYF2 in PD remains controversial,5 the GIGYF2 p.Arg610Gly mutation, which is situated in the GYF domain of the protein that is thought to possess ligand-binding properties,6 was shown to be highly conserved across different species in both GIGYF1 and GIGYF2 proteins (Figure 1), and was predicted to disrupt the binding between the protein’s GYF domain and its interacting ligands (data not shown). The exon containing the p.Arg610Gly mutation was then sequenced in 107 Spanish PD patients, yielding no additional pathogenic mutation carriers; and the entire coding region of GIGYF2 was sequenced in 45 Spanish PD patients (age at onset ranged from 61 to 81 years), leading to the identification of one novel mutation, p.Lys1006Gln_insQ, which is probably non-pathogenic as it lies within a highly polymorphic polyglutamine repeat, along with other already-reported, non-pathogenic mutations. All mutations identified were later tested in neurologically normal individuals and excluded from being risk factors for PD (Table 2a–c).
Among the other SNVs identified, we suspected that only the mutation within MTHFD1L, which has been associated with an increased risk of late-onset Alzheimer disease7 and neural tube defects,8 may have role in the phenotypic expression of our family. The MTHFD1L p.Ala844Ser mutation was then investigated in 107 Spanish PD patients and 105 Spanish patients with late-onset Alzheimer disease. No additional mutation carrier was identified, suggesting that it has no implications in the pathophysiology of PD and AD. The remaining SNVs identified were localized in FN1 and C1QL2 genes, respectively, and as such are unlikely to have a role in the pathogenesis of PD: FN1 encoding for fibronectin is responsible for glomerulopathy with fibronectin deposits in humans9 and has been involved in cell adhesion and migration processes, including embryogenesis, wound healing, blood coagulation, host defense and metastasis;10 C1QL2, which is not expressed in brain tissues,11 belongs to a large family of multimeric secreted glycoproteins.
Given the physiological role of GIGYF2 in the regulation of vesicular transport and insulin/insulin like growth factor 1 (IGF-1) signaling in the central nervous system,12, 13 the role of insulin in the regulation of brain dopaminergic activity14 and the identification of elevated levels of IGF-1 and IGF-binding proteins in the serum and cerebrospinal fluid of patients with PD,15 we hypothesize that aberrations in proteins involved in the insulin/IGF-1 signaling pathway, including GIGYF2, may be the key players in the pathogenesis of LOPD. The fact that most biological functions of IGF-1, which acts as a homeostatic modulator for normal brain functionality and synaptic plasticity, are mediated by the IGF-1 receptor16 and that GIGYF2 has been shown to play role in the regulation of IGF-1 receptor trafficking in specific, mammalian, neuronal populations, including hippocampal pyramidal neurons also supports this hypothesis.17 Even though high prevalence of insulin resistance has been reported in patients with PD18 and different studies have revealed an important role of insulin in normal memory function and learning ability,19 a possible role of the MTHFD1L gene in the cognitive dysfunction of our reported family cannot be ruled out. However, this coupled with the fact that overexpression of GIGYF2 has not only been shown to correlate with an increased neuronal apoptosis but also to diminish cognitive function17 may suggest that the cognitive impairment seen in our family may be due to a possible insulin dysregulation caused by GIGYF2 genetic variability identified in this study. Because dysregulation of insulin may predispose neurodegenerative disease late in life,19 GIGYF2 mutation carriers may not develop the full parkinsonian symptoms until an advanced age as occurred in our reported family.
In summary, our study and others suggest that GIGYF2 genetic variability may be, although rare, a cause of LOPD. Although there are still many challenges to be met, this study adds insights into the contribution of GIGYF2 to the pathogenesis of PD and suggests insulin dysregulation as a disease-specific mechanism for both PD and cognitive dysfunction.
References
Bonifati, V. Genetics of Parkinson's disease—state of the art, 2013. Parkinsonism Relat. Disord. 20 (Suppl 1), S23–S28 (2013).
Louis, E. D., Rios, E., Applegate, L. M., Hernandez, N. C. & Andrews, H. F. Jaw tremor: prevalence and clinical correlates in three essential tremor case samples. Mov. Disord. 21, 1872–1878 (2006).
Krebs, C. E., Karkheiran, S., Powell, J. C., Cao, M., Makarov, V., Darvish, H. et al. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum. Mutat. 34, 1200–1207 (2013).
Exome Variant Server NHLBI GO Exome Sequencing Project (ESP). Seattle, WA, USA, (April 2015; URL http://evs.gs.washington.edu/EVS/ (2015).
Tan, E. K. & Schapira, A. H. Summary of GIGYF2 studies in Parkinson's disease: the burden of proof. Eur. J. Neurol. 17, 175–176 (2009).
Freund, C., Dotsch, V., Nishizawa, K., Reinherz, E. L. & Wagner, G. The GYF domain is a novel structural fold that is involved in lymphoid signaling through proline-rich sequences. Nat. Struct. Biol. 6, 656–660 (1999).
Naj, A. C., Beecham, G. W., Martin, E. R., Gallins, P. J., Powell, E. H., Konidari, I. et al. Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS Genet. 6, e1001130 (2010).
Parle-McDermott, A., Pangilinan, F., O'Brien, K. K., Mills, J. L., Magee, A. M., Troendle, J. et al. A common variant in MTHFD1L is associated with neural tube defects and mRNA splicing efficiency. Hum. Mutat. 30, 1650–1656 (2009).
Castelletti, F., Donadelli, R., Banterla, F., Hildebrandt, F., Zipfel, P. F., Bresin, E. et al. Mutations in FN1 cause glomerulopathy with fibronectin deposits. Proc. Natl Acad. Sci. USA 105, 2538–2543 (2008).
Steffens, S., Schrader, A. J., Vetter, G., Eggers, H., Blasig, H., Becker, J. et al. Fibronectin 1 protein expression in clear cell renal cell carcinoma. Oncol. Lett. 3, 787–790 (2012).
Wong, G. W., Krawczyk, S. A., Kitidis-Mitrokostas, C., Revett, T., Gimeno, R. & Lodish, H. F. Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem. J. 416, 161–177 (2008).
Nunez, A., Carro, E. & Torres-Aleman, I. Insulin-like growth factor I modifies electrophysiological properties of rat brain stem neurons. J. Neurophysiol. 89, 3008–3017 (2003).
O'Kusky, J. R., Ye, P. & D'Ercole, A. J. Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J. Neurosci. 20, 8435–8442 (2000).
Liu, Z., Wang, Y., Zhao, W., Ding, J., Mei, Z., Guo, L. et al. Peptide derived from insulin with regulatory activity of dopamine transporter. Neuropharmacology 41, 464–471 (2001).
Mashayekhi, F., Mirzajani, E., Naji, M. & Azari, M. Expression of insulin-like growth factor-1 and insulin-like growth factor binding proteins in the serum and cerebrospinal fluid of patients with Parkinson's disease. J. Clin. Neurosci. 17, 623–627 (2010).
Aleman, A. & Torres-Aleman, I. Circulating insulin-like growth factor I and cognitive function: neuromodulation throughout the lifespan. Prog. Neurobiol. 89, 256–265 (2009).
Xie, J., Wei, Q., Deng, H., Li, G., Ma, L. & Zeng, H. Negative regulation of Grb10 Interacting GYF Protein 2 on insulin-like growth factor-1 receptor signaling pathway caused diabetic mice cognitive impairment. PLoS ONE. 9, e108559 (2014).
Sandyk, R. The relationship between diabetes mellitus and Parkinson's disease. Int. J. Neurosci. 69, 125–130 (1993).
Craft, S. & Watson, G. S. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 3, 169–178 (2004).
Acknowledgements
We thank patients, relatives and all participants for their contribution to this research. Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS079388 (CP-R).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Ruiz-Martinez, J., Krebs, C., Makarov, V. et al. GIGYF2 mutation in late-onset Parkinson’s disease with cognitive impairment. J Hum Genet 60, 637–640 (2015). https://doi.org/10.1038/jhg.2015.69
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.69
