
Abstract
Myofibrillar myopathy (MFM) is a group of chronic muscular disorders that show the focal dissolution of myofibrils and accumulation of degradation products. The major genetic basis of MFMs is unknown. In 1993, our group reported a Japanese family with dominantly inherited cytoplasmic body myopathy, which is now included in MFM, characterized by late-onset chronic progressive distal muscle weakness and early respiratory failure. In this study, we performed linkage analysis and exome sequencing on these patients and identified a novel c.90263G>T mutation in the TTN gene (NM_001256850). During the course of our study, another groups reported three mutations in TTN in patients with hereditary myopathy with early respiratory failure (HMERF, MIM #603689), which is characterized by overlapping pathologic findings with MFMs. Our patients were clinically compatible with HMERF. The mutation identified in this study and the three mutations in patients with HMERF were located on the A-band domain of titin, suggesting a strong relationship between mutations in the A-band domain of titin and HMERF. Mutation screening of TTN has been rarely carried out because of its huge size, consisting of 363 exons. It is possible that focused analysis of TTN may detect more mutations in patients with MFMs, especially in those with early respiratory failure.
Similar content being viewed by others


Introduction
Myofibrillar myopathies (MFMs) were proposed in 1996 as a group of chronic muscular disorders characterized by common morphologic features observed on muscle histology, which showed the focal dissolution of myofibrils followed by the accumulation of products of the degradative process.1 The clinical phenotype of MFM is characterized by slowly progressive muscle weakness that can involve proximal or distal muscles, with onset in adulthood in most cases. However, other phenotypes are highly variable. Although 20% of patients with MFMs have been revealed to have mutations in DES, CRYAB, MYOT, LDB (ZASP), FLNC or BAG3, the major genetic basis of MFMs remains to be elucidated.
Respiratory weakness is one of the symptoms of MFMs. The early or initial presentation of respiratory failure is not a common manifestation of MFMs as a whole, and there are limited reports regarding a fraction of patients with DES,2 MYOT3 or CRYAB4 mutation. In 1993, our group reported a Japanese family with dominantly inherited cytoplasmic body (CB) myopathy,5 which is now included in MFM. Currently, this family includes 20 patients in five successive generations who show almost homogeneous clinical features characterized by chronic progressive distal muscle weakness and early respiratory failure. However, the underlying genetic etiology in this family was unknown. The aim of this study was to determine the genetic cause in this family. To identify the responsible genetic mutation, we performed linkage analysis and whole-exome sequencing.
Materials and methods
This study was approved by the Ethics Committee of the Tohoku University School of Medicine, and all individuals gave their informed consent before their inclusion in the study.
Clinical information on the family
This family includes 20 patients (13 males and 7 females) in five successive generations (Figure 1). The family is of Japanese ancestry, and no consanguineous or international mating was found. Of all patients, seven underwent a muscle biopsy, and two were autopsied. All of the histological findings were compatible with MFM (see clinical data).

Family pedigree. Filled-in symbols indicate individuals with MFM. Empty symbols indicate unaffected individuals. A star and asterisk indicate autopsy-proven and muscle biopsy-proven cases, respectively. (A–J) indicates individuals whose DNA was used for this study.
The age of onset ranged from 27–45 years. The most common presenting symptom was foot drop. At the initial evaluations, muscle weakness was primarily distributed in the ankle dorsiflexors and finger extensors. The patients were generally built and showed no other extramuscular abnormalities. In addition to this chronic progressive distal muscle weakness, respiratory distress occurred between 0 and 7 years from the initial onset (average 3.8 years) in seven patients (IV-9, V-2, A, B, E, H, and J) with adequate clinical information. Two patients who had not had any respiratory care died of respiratory failure approximately a decade from the initial onset. The other patients have been alive for more than 10 years (maximum 18 years) but require nocturnal non-invasive positive pressure ventilation. They were 37–58 years of age as of 2012 and able to walk independently with or without a simple walking aid. Although the time at which patients recognized dysphagia or dysarthria varied between 1 to more than 10 years from the initial onset, decreased bulbar functions had been noted at the initial evaluation in most cases. Cardiac function was normally maintained in all patients of the family.
Clinical data
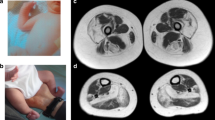
The level of serum creatine kinase was normal or mildly elevated. Electromyography of affected muscles showed a chronic myogenic pattern, and the nerve conduction study did not suggest any neuropathic involvement. Muscle imaging showed focal atrophy in the tibialis anterior, tibialis posterior, extensor hallucis and digitorum longus, peroneal and semitendinosus muscle on initial assessment (Figure 2A), and atrophy became clear in cervical muscles, shoulder girdles, intercostals and proximal limb muscles in the following several years. Upon muscle biopsy, the most common finding was numerous cytoplasmic bodies (CBs), which were found on 7.3% of myofibers in the tibialis anterior of individual E (Figure 2B (a–c)) and 50–80% of intercostals in other cases.5 Other nonspecific findings were increased variability in the size of myofibers, central nuclei and rimmed vacuoles observed on a few fibers. No strong immunoreaction of desmin was seen in the CBs (Figure 2B (d, e)). An electron microscope examination showed that the regular sarcoplasmic pattern was replaced by abnormal fine filamentous structures, which seemed to attach to the Z-band. CBs were also found in almost all skeletal muscles and some smooth muscles in autopsied cases.5 Cardiac myofibers also contained numerous CBs in one of the autopsied cases (V-2),5 although the patient did not present any cardiac complication. The sequence analysis of the coding regions and flanking introns of DES and MYOT showed no pathogenic mutation in individual E. An array comparative genomic hybridization performed with the Agilent SurePrint G3 Human CGH 1M microarray format in individual A did not reveal any aberrations of genomic copy number.

Family clinical data. (A) Muscle computed tomography of affected lower extremity. The imaging in the initial assessment of individual A showed symmetrical atrophy and fatty replacement of the semitendinosus in the proximal lower extremities (left) and the tibialis anterior, tibialis posterior, extensor hallucis and digitorum longus, and peroneal muscle in the distal (right) lower extremities. (B) Pathology of muscle biopsy. Hematoxylin-eosin (a), Gomori-trichrome (b) and NADH (nicotinamide adenine dinucleotide)-tetrazolium reductase (c) staining of the muscle biopsy sample from the tibialis anterior of individual E are shown. CBs are indicated by arrows. CBs were round or oval, 5–10 μm in diameter and predominantly located in the periphery of type 1 fibers, which stained eosinophilic with hematoxylin-eosin and blue–purple with Gomori-trichrome. NADH-tetrazolium reductase staining showed disorganization of the myofibrillar network. Immunostaining for desmin (d) and Gomori-trichrome staining (e) are serial sections of the muscle biopsy from individual E. Stars indicate corresponding fibers. No strong immunoreaction of desmin was seen in the CBs. Scale bars=100 μm
Linkage analysis
DNA was extracted by standard methods. Linkage analysis was performed on nine family members (A–I in Figure 1; four of them were affected, and the others were unaffected) through genotyping using an Illumina Human Omni 2.5 BeadChip (Illumina, San Diego, CA, USA). We chose single-nucleotide polymorphisms (SNPs) that satisfied all of the following criteria: (1) autosomal SNPs whose allele frequencies were available from the HapMap project (http://hapmap.ncbi.nlm.nih.gov/), (2) SNPs that were not monomorphic among members and (3) SNPs that were not in strong linkage disequilibrium with neighboring SNPs (r2 values <0.9). Then, we selected the first five SNPs from each position of integer genetic distance from SNPs that met the above criteria for the initial analysis. The details were as follows; we chose a SNP closest to 0 cM and the neighboring four SNPs. If the genetic distance of a SNP was the same as that of the next SNP, we considered the genomic position to determine their order. We repeated this process at 1 cM, 2 cM and so on.
We performed a multipoint linkage analysis of the data set (17 613 SNPs) using MERLIN6 1.1.2 under the autosomal dominant mode with the following parameters: 0.0001 for disease allele frequency, 1.00 for individuals heterozygous and homozygous for the disease allele and 0.00 for individuals homozygous for the alternative allele. After this first analysis, a second analysis was performed with all SNPs fulfilling the above criteria around the peaks identified in the first analysis.
Exome sequencing
Exome sequencing was performed on seven family members in three generations (A–E, H and I in Figure 1), four of whom were affected. Exon capture was performed with the SureSelect Human All Exon kit v2 (individuals E, H and I) or v4 (A–D) (Agilent Technologies, Santa Clara, CA, USA). Exon libraries were sequenced with the Illumina Hiseq 2000 platform according to the manufacturer’s instructions (Illumina). Paired 101-base pair reads were aligned to the reference human genome (UCSChg19) using the Burrows-Wheeler Alignment tool.7 Likely PCR duplicates were removed with the Picard program (http://picard.sourceforge.net/). Single-nucleotide variants and indels were identified using the Genome Analysis Tool Kit (GATK) v1.5 software.8 SNVs and indels were annotated against the RefSeq database and dbSNP135 with the ANNOVAR program.9 We used the PolyPhen2 polymorphism phenotyping software tool10 to predict the functional effects of mutations.
Sanger sequencing
To confirm that mutations identified by exome sequencing segregated with the disease, we performed direct sequencing. PCR was performed with the primers shown in Supplementary Table 1. PCR products were purified with a MultiScreen PCR plate (Millipore, Billerica, MA, USA) and sequenced using BigDye terminator v1.1 and a 3500xL genetic analyzer (Applied Biosystems, Carlsbad, CA, USA).
Results
Linkage analysis
The first linkage analysis identified five regions across autosomes with a logarithm of odds (LOD) score greater than 2 (Figure 3). Of the five regions, two were on chromosome 2 (from 167 cM to 168 cM, with a maximum LOD score of 2.46 and from 182 cM to 185 cM, with a maximum LOD score of 2.71), the other two were on chromosome 8 (from 27 cM to 34 cM, with a maximum LOD score of 2.71 and at 61 cM, with a maximum LOD score of 2.03), and one was on chromosome 17 (at 5 cM, with a maximum LOD score of 2.53). In the second detailed linkage analysis, these peaks were determined to range from 167.49 cM at rs4233674 at position 159 058 679 to 168.19 cM at rs7598162 at position 160 935 582, and from 181.23 cM at rs4402725 at position 174 893 412 to 187.05 cM at rs7420169 at position 182 548 671 on chromosome 2; from 26.42 cM at rs2736043 at position 15 713 330 to 34.88 cM at rs9325871 at position 20 391 160, and from 61.02 cM at rs6999814 at position 41 660 854 to 62.32 cM at rs10957281 at position 49 769 454 on chromosome 8; and from 4.7 cM at rs11078552 at position 1 550 848 to 5.45 cM at rs1057355 at position 1 657 899 on chromosome 17. Haplotypes shared by affected individuals in these regions were confirmed by visual inspection. There were a few incompatible SNPs in these regions, presumably due to genotyping error.

Linkage analysis. Linkage analysis was performed on nine family members (four of them were affected, the others were unaffected) using an Illumina Human Omni 2.5 BeadChip. Five regions with an LOD score greater than 2 (indicated by bar) were identified. A full color version of this figure is available at the Journal of Human Genetics journal online.
Exome sequencing and segregation analysis
In exome sequencing, an average of 215 million reads enriched by SureSelect v4 (SSv4) and 319 million reads enriched by SureSelect v2 (SSv2) were generated, and 99% of reads were mapped to the reference genome by Burrows-Wheeler Alignment tool. An average of 57% (SSv4) and 61% (SSv2) of those reads were duplicated and removed, and an average of 80% (SSv4) and 66% (SSv2) of mapped reads without duplicates were in target regions. The average coverage of each exome was 163-fold (SSv4) and 130-fold (SSv2). An average of 85% (SSv4) and 69% (SSv2) of target regions were covered at least 50-fold (Supplementary Table 2). On average, 10 133 SNVs or indels, which are located within coding exons or splice sites, were identified per individual (Table 1). A total of 64 variants were common among patients and not present in unaffected individuals, and 32 of those were left after excluding synonymous SNVs. In these variants, only the heterozygous mutation c.90263G>T (NM_001256850) at position 179 410 777 of chromosome 2, which was predicted to p.W30088L in TTN, was novel (that is, not present in dbSNP v135 or 1000 genomes). Polyphen2 predicted this mutation as probably damaging. This mutation was located in a candidate region suggested by the linkage analysis in the present study. The other variants were registered with dbSNP135, and the allele frequencies, except for one SNV, rs138183879, in IKBKB, ranged from 0.0023 to 0.62. These values were not compatible with the assumption that MFM was a rare disease and showed complete penetrance in this family. The allele frequency of rs138183879 was not available in dbSNP135, and this SNV was in the candidate region on chromosome 8 based on linkage analysis.
We then performed a segregation analysis on the two candidates, the novel mutation c.90263G>T in TTN and rs138183879 in IKBKB, through Sanger sequencing in 10 family members (A–J in Figure 1; Figure 4a). The rs138183879 SNP was not found in individual J, that is, it was not segregated with the disease in this family. In contrast, the novel mutation c.90263G>T in TTN was detected in all patients (n=5) and not detected in any of the unaffected family members (n=5) or 191 ethnically matched control subjects (382 chromosomes). These results suggested that this rare mutation in TTN segregated with the disease in this family.

Identified mutations by exome sequencing. (a) We performed segregation analysis of two candidates. (b) The identified TTN mutation and its conservation among species. Sanger sequencing confirmed the heterozygous G to T substitution (indicated by the arrow) at the position chr2;179 410 777, which corresponds to c.90263G>T in exon 293 (NM_001256850.1). The substitution leads to p.W30088L (NP_001243779.1), and this amino acid is conserved among species.
Discussion
In this study, we found that a novel missense mutation in TTN segregated with MFM in a large Japanese family. The identified c.90263G>T mutation in TTN (NM_001256850) was considered to be the genetic cause of MFM in our family, because (1) exome sequencing revealed that this was the best candidate mutation after filtering SNPs and indels, (2) this mutation is located in a region on chromosome 2 shared by affected family members, (3) the segregation with MFM was confirmed by Sanger sequencing, (4) this mutation was not detected in 191 control individuals, (5) this mutation was predicted to alter highly conserved amino acids (Figure 4b) and (6) TTN encodes a Z-disc-binding molecule called titin, which is similar to all of the previously identified causative genes for MFMs, which also encode Z-disc-associated molecules.
Recently, three mutations in TTN have been reported as the causes of hereditary myopathy with early respiratory failure (HMERF, MIM #603689),11, 12, 13, 14, 15, 16 which has similar muscle pathology to MFMs. The identified novel missense mutation c.90263G>T in our study was located on the same exon as recently reported HMERF mutations: c.90272C>T in a Portuguese family16 and c.90315T>C in Swedish and English families14, 15 (Table 2). This finding suggests the possibility that our family can be recognized as having HMERF from a clinical aspect.
Compared with symptoms described in the past three reports on HMERF (also see Table 2), our patients have common features, such as autosomal dominant inheritance, early respiratory failure, the absence of clinically apparent cardiomyopathy, normal to mild elevation of serum CK and histological findings compatible with MFM. Early involvement of the tibialis anterior is also common, except for the Portuguese family, who reported isolated respiratory insufficiency and a milder presentation of HMERF. Thus, our family shares major clinical manifestations with patients with HMERF, suggesting that the identified mutation is novel for MFM and HMERF.
To date, mutations in TTN have been identified in skeletal myopathy and cardiomyopathy.17, 18 The relationship between the variant positions on TTN and phenotypes accompanied by skeletal or respiratory muscle involvement is summarized in Table 2. Titin is a large protein (4.20 MDa) that extends from the Z-disk to the M-line within the sarcomere, and it is composed of four major domains: Z-disc, I-band, A-band and M-line (Figure 5). All four HMERF mutations detected by other groups and our study were consistently located in the A-band domain, while mutations in tibial muscular dystrophy (TMD) (MIM #600334),19, 20, 21, 22, 23, 24 limb-girdle muscular dystrophy type 2J (LGMD2J) (#608807)19, 25 and early-onset myopathy with fatal cardiomyopathy (#611705)26 were located in the M-line domain. HMERF and TMD have some common clinical characteristics, such as autosomal dominant inheritance with onset in adulthood and strong involvement of the tibialis anterior muscle. In contrast, one of the distinctive features of TMD is that early respiratory failure has not been observed in patients with TMD. Histological findings of TMD usually do not include CBs but show nonspecific dystrophic change. The underlying pathogenic processes explaining why mutations on these neighboring domains share some similarities but also some differences are unknown.

Structure of titin and mutation distribution in the A-band domain. Human TTN was mapped to 2q31.2. TTN is 294 kb and is composed of 363 exons that code for a maximum of 38 138 amino-acid residues and a 4.20-MDa protein32 called titin. Titin is expressed in the cardiac and skeletal muscles and spans half the sarcomere, with its N-terminal at the Z-disc and the C-terminal at the M-line.33 Titin is composed of four major domains: Z-disc, I-band, A-band and M-line. I-band regions of titin are thought to make elastic connections between the thick filament (that is, myosin filament) and the Z-disc within the sarcomere, whereas the A-band domain of titin seems to be bound to the thick filament, where it may regulate filament length and assembly.34 The gray and white ellipses indicate an Ig-like domain and fibronectin type 3 domain, respectively. Our mutation (p.W30088L) and the neighboring two mutations (that is, p.C30071R and p.P30091L) were all located in the 6th Fn3 domain in the 10th domain of large super-repeats. A full color version of this figure is available at the Journal of Human Genetics journal online.
Three of four HMERF mutations in the A-band domain are located in the fibronectin type 3 and Ig-like (Fn3/Ig) domain, and one of four HMERF mutations is located in the kinase domain (Table 2, also see Figure 5). The missense mutation c.97348C>T in the kinase domain was the first reported HMERF mutation. It has been shown that the kinase domain has an important role in controlling muscle gene expression and protein turnover via the neighbor of BRCA1 gene-1-muscle-specific RING finger protein-serum response transcription factor pathway.13 Moreover, the Fn3/Ig domain is composed of two types of super-repeats: six consecutive copies of 7-domain super-repeat at the N-terminus and 11 consecutive copies of 11-domain super-repeat at the C-terminus.27, 28, 29 These super-repeats are highly conserved among species and muscles. Our identified mutation (c.90263G>T) and the neighboring two mutations (that is, c.90272C>T and c.90315T>C shown in Table 2) were all located on the 6th Fn3 domain in the 10th copy of 11-domain super-repeat (that is, A150 domain30) (Figure 5). Although some Fn3 domains are proposed to be the putative binding site for myosin,31 the role with the majority of Fn3 domains, how it supports the structure of each repeat architecture, and the identity of its binding partner have not been fully elucidated. Our findings suggested that the Fn3 domain, in which mutations clustered, has critical roles in the pathogenesis of HMERF, although detailed mechanisms of pathogenesis remain unknown.
In conclusion, we have identified a novel disease-causing mutation in TTN in a family with MFM that was clinically compatible with HMERF. Because of its large size, global mutation screening of TTN has been difficult. Mutations in TTN may be detected by massively parallel sequencing in more patients with MFMs, especially in patients with early respiratory failure. Further studies are needed to understand the genotype–phenotype correlations in patients with mutations in TTN and the molecular function of titin.
References
Nakano, S., Engel, A. G., Waclawik, A. J., Emslie-Smith, A. M. & Busis, N. A. Myofibrillar myopathy with abnormal foci of desmin positivity. I. Light and electron microscopy analysis of 10 cases. J. Neuropathol. Exp. Neurol. 55, 549–562 (1996).
Olive, M., Odgerel, Z., Martinez, A., Poza, J. J., Bragado, F. G., Zabalza, R. J. et al. Clinical and myopathological evaluation of early- and late-onset subtypes of myofibrillar myopathy. Neuromuscul. Disord. 21, 533–542 (2011).
Olive, M., Goldfarb, L. G., Shatunov, A., Fischer, D. & Ferrer, I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain 128, 2315–2326 (2005).
Selcen, D. & Engel, A. G. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann. Neurol. 54, 804–810 (2003).
Abe, K., Kobayashi, K., Chida, K., Kimura, N. & Kogure, K. Dominantly inherited cytoplasmic body myopathy in a Japanese kindred. Tohoku. J. Exp. Med. 170, 261–272 (1993).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome. Res. 20, 1297–1303 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data.. Nucleic Acids Res. 38, e164 (2010).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Nicolao, P., Xiang, F., Gunnarsson, L. G., Giometto, B., Edstrom, L., Anvret, M. et al. Autosomal dominant myopathy with proximal weakness and early respiratory muscle involvement maps to chromosome 2q. Am. J. Hum. Genet. 64, 788–792 (1999).
Edstrom, L., Thornell, L. E., Albo, J., Landin, S. & Samuelsson, M. Myopathy with respiratory failure and typical myofibrillar lesions. J. Neurol. Sci. 96, 211–228 (1990).
Lange, S., Xiang, F., Yakovenko, A., Vihola, A., Hackman, P., Rostkova, E. et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science 308, 1599–1603 (2005).
Ohlsson, M., Hedberg, C., Bradvik, B., Lindberg, C., Tajsharghi, H., Danielsson, O. et al. Hereditary myopathy with early respiratory failure associated with a mutation in A-band titin. Brain 135, 1682–1694 (2012).
Pfeffer, G., Elliott, H. R., Griffin, H., Barresi, R., Miller, J., Marsh, J. et al. Titin mutation segregates with hereditary myopathy with early respiratory failure. Brain 135, 1695–1713 (2012).
Vasli, N., Bohm, J., Le Gras, S., Muller, J., Pizot, C., Jost, B. et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta. Neuropathol. 124, 273–283 (2012).
Kontrogianni-Konstantopoulos, A., Ackermann, M. A., Bowman, A. L., Yap, S. V. & Bloch, R. J. Muscle giants: molecular scaffolds in sarcomerogenesis. Physiol. Rev. 89, 1217–1267 (2009).
Ottenheijm, C. A. & Granzier, H. Role of titin in skeletal muscle function and disease. Adv. Exp. Med. Biol. 682, 105–122 (2010).
Hackman, P., Vihola, A., Haravuori, H., Marchand, S., Sarparanta, J., De Seze, J. et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am. J. Hum. Genet. 71, 492–500 (2002).
Udd, B., Partanen, J., Halonen, P., Falck, B., Hakamies, L., Heikkila, H. et al. Tibial muscular dystrophy. Late adult-onset distal myopathy in 66 Finnish patients. Arch. Neurol. 50, 604–608 (1993).
de Seze, J., Udd, B., Haravuori, H., Sablonniere, B., Maurage, C. A., Hurtevent, J. F. et al. The first European family with tibial muscular dystrophy outside the Finnish population. Neurology 51, 1746–1748 (1998).
Van den Bergh, P. Y., Bouquiaux, O., Verellen, C., Marchand, S., Richard, I., Hackman, P. et al. Tibial muscular dystrophy in a Belgian family. Ann. Neurol. 54, 248–251 (2003).
Hackman, P., Marchand, S., Sarparanta, J., Vihola, A., Penisson-Besnier, I., Eymard, B. et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul. Disord. 18, 922–928 (2008).
Pollazzon, M., Suominen, T., Penttila, S., Malandrini, A., Carluccio, M. A., Mondelli, M. et al. The first Italian family with tibial muscular dystrophy caused by a novel titin mutation. J. Neurol. 257, 575–579 (2010).
Udd, B., Rapola, J., Nokelainen, P., Arikawa, E. & Somer, H. Nonvacuolar myopathy in a large family with both late adult onset distal myopathy and severe proximal muscular dystrophy. J. Neurol. Sci. 113, 214–221 (1992).
Carmignac, V., Salih, M. A., Quijano-Roy, S., Marchand, S., Al Rayess, M. M., Mukhtar, M. M. et al. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann. Neurol. 61, 340–351 (2007).
Labeit, S., Barlow, D. P., Gautel, M., Gibson, T., Holt, J., Hsieh, C. L. et al. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature 345, 273–276 (1990).
Labeit, S. & Kolmerer, B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270, 293–296 (1995).
Tskhovrebova, L., Walker, M. L., Grossmann, J. G., Khan, G. N., Baron, A. & Trinick, J. Shape and flexibility in the titin 11-domain super-repeat. J. Mol. Biol. 397, 1092–1105 (2010).
Bucher, R. M., Svergun, D. I., Muhle-Goll, C. & Mayans, O. The structure of the FnIII Tandem A77-A78 points to a periodically conserved architecture in the myosin-binding region of titin. J. Mol. Biol. 401, 843–853 (2010).
Muhle-Goll, C., Habeck, M., Cazorla, O., Nilges, M., Labeit, S. & Granzier, H. Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1–a possible role in the Frank-Starling mechanism of the heart. J. Mol. Biol. 313, 431–447 (2001).
Bang, M. L., Centner, T., Fornoff, F., Geach, A. J., Gotthardt, M., McNabb, M. et al. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ. Res. 89, 1065–1072 (2001).
Maruyama, K., Yoshioka, T., Higuchi, H., Ohashi, K., Kimura, S. & Natori, R. Connectin filaments link thick filaments and Z lines in frog skeletal muscle as revealed by immunoelectron microscopy. J. Cell. Biol. 101, 2167–2172 (1985).
Guo, W., Bharmal, S. J., Esbona, K. & Greaser, M. L. Titin diversity–alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 753675 (2010).
Acknowledgements
We thank the patients and their family. We are grateful to Yoko Tateda, Kumi Kato, Naoko Shimakura, Risa Ando, Riyo Takahashi, Miyuki Tsuda, Nozomi Koshita, Mami Kikuchi and Kiyotaka Kuroda for their technical assistance. We also acknowledge the support of the Biomedical Research Core of Tohoku University Graduate School of Medicine. This work was supported by a grant of Research on Applying Health Technology provided by the Ministry of Health, Labor and Welfare to YM, an Intramural Research Grant (23-5) for Neurological and Psychiatric Disorders of NCNP and JSPS KAKENHI Grant number 24659421.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Izumi, R., Niihori, T., Aoki, Y. et al. Exome sequencing identifies a novel TTN mutation in a family with hereditary myopathy with early respiratory failure. J Hum Genet 58, 259–266 (2013). https://doi.org/10.1038/jhg.2013.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.9
Keywords
This article is cited by
-
A novel deletion in the C-terminal region of HSPB8 in a family with rimmed vacuolar myopathy
Journal of Human Genetics (2021)
-
Expanding the importance of HMERF titinopathy: new mutations and clinical aspects
Journal of Neurology (2019)
-
Myofibrillar myopathy in the genomic context
Journal of Applied Genetics (2018)
-
Performance comparison of two commercial human whole-exome capture systems on formalin-fixed paraffin-embedded lung adenocarcinoma samples
BMC Cancer (2016)
-
Diagnosis of muscle diseases presenting with early respiratory failure
Journal of Neurology (2015)
