
Abstract
Fabry disease (FD) is an X-linked lysosomal storage disorder caused by a deficiency of α-galactosidase A (GLA) activity. Enzyme replacement therapy (ERT) for FD is available, and newborn mass screening for FD is being implemented. Here, we undertook a pilot study of newborn mass screening for FD in Japan. GLA activity in dried blood spots was measured using a fluorescence assay and confirmed by measurement of GLA activity in white blood cells (WBCs) in infants with abnormally low GLA activity. This was followed up by genetic testing. A total of 21 170 neonates were enrolled in the study. Of these, seven (five boys, two girls) had low GLA activities, which were verified by the WBC GLA activity assay. Thus, the initial fluorescence assay was suitable for newborn mass screening for FD. Pathogenic mutations of the GLA gene, that is, V199M and IVS4+919G>A, were found in two boys and one boy, respectively. Functional mutations, E66Q and c.−10C>T: g.1170C>T, were found in two boys and one girl, respectively. The prevalence of test-positive newborns was 1/3024, while that of those with a pathogenic mutation was 1/7057. The numbers are higher than those previously anticipated. Standardized management for FD found during newborn mass screening, including an ERT regimen, remains to be established.
Similar content being viewed by others

Introduction
Fabry disease (FD: MIM 301500) is an X-linked inherited lysosomal storage disorder caused by a deficiency in α-galactosidase A (GLA) activity.1 This deficiency leads to the accumulation of globotriaosylceramide in different tissues and can cause progressive malfunctions in systemic organs, such as the skin, eyes, kidneys, ears, lungs, heart and brain.1, 2, 3
Male patients with classic early-onset FD usually have very low GLA activity and are generally asymptomatic in early childhood (onset symptoms are reported at a mean age of 9 years).4, 5 The major clinical symptoms of classical early-onset FD include pain in the peripheral extremities, angiokeratoma, hypohidrosis, corneal opacity, and renal, cardiac and cerebrovascular diseases.1 In contrast, patients with late-onset FD exhibit residual GLA activity and milder clinical manifestations than those with classical early-onset FD.1 Heterozygous females with FD have wide clinical manifestation spectra, ranging from asymptomatic to severely affected.6
In 2001, in the USA and EU, enzyme replacement therapy (ERT) was approved for the treatment of FD; Japan began using ERT in 2004. In all three of these regions, ERT has been shown to be effective in alleviating many of the signs and symptoms of the disease and in slowing or even reversing disease progression.7, 8, 9 Several studies have demonstrated that ERT must be administered before the occurrence of renal or cardiac failure in order to achieve optimal results.10, 11, 12 As the importance of early treatment is now generally recognized, newborn mass screening for FD is being implemented in several countries. Such newborn mass screening, however, had not yet been undertaken in Japan; hence, the prevalence and genotypes of FD in association with GLA activity have not been studied in the Japanese population.
In this work, we present the results of a pilot study for newborn mass screening for FD in Fukuoka City and its vicinity in Japan.
Subjects and methods
Subjects
This study was conducted from April 2007 to April 2010 in Fukuoka City and its vicinity in Japan. Among newborns who took the conventional newborn mass screening during the study period, only newborns whose parent gave their written consent to participate were enrolled in the study. Conventional newborn mass screening has taken place as a local administrative service nationwide in Japan to screen six disorders: cretinism, congenital adrenal hypertrophy, galactosemia, phenylketonuria, homocystinemia and maple syrup urine disease. Virtually all newborns born in Japan take this conventional newborn screening. The dried blood spots that remained after completion of the conventional newborn mass screening were used for this study. The sex of newborns enrolled in the study was judged according to information written on the filter papers; this information was not available for some of the enrolled newborns because of incomplete descriptions.
Methods
Venous blood was collected from neonates on days 4–6 after birth, transferred to filter paper, and dried at room temperature for the conventional newborn screening and this study. A small circle of 3 mm in diameter was punched out of the dried blood spot and used for mass screening for FD.3, 13, 14
In the mass-screening study, GLA activity was determined using a fluorescent substrate, as described previously.3, 13, 14 In brief, 40 μl McIlvan buffer (0.1 M citrate: 0.2 M NaH2PO4, 36.8:63.2 v/v, pH 6.0) was added to each well of 96-microwell plates. Punched dried blood spots were added to the buffer and processed for extraction at room temperature for 2 h. Aliquots of 30 μl of blood extract were transferred to new 96-microwell plates. An aliquot of 100 μl of the reaction mixture (3.5 mM 4-methylumbelliferone (4-MU) galactosylpyranoside, 100 mM citrate, 200 mM phosphate and 100 mM N-acetylgalactosamine, pH 4.4) was added to each well of the 96-microwell plates and incubated at 37 °C for 24 h. The reaction was terminated with 150 μl termination solution (300 mM glycine, NaOH, pH 10.6) immediately after the reaction. The fluorescence intensity from 4-MUs in the wells was measured with a fluorescence plate reader (Bio-Tek, Winooski, VT, USA) at 450 nm. One unit (AgalU) of enzymatic activity was equal to 0.34 pmol of 4-methylumbelliferyl-D-galactopyranoside cleaved per hour per disc. When GLA activity was identified as being abnormally low (i.e., less than the cutoff value of 20 AgalU), a second measurement was taken 2 or 3 weeks later to verify the initial measurement.14 Newborns whose GLA activity had been verified as <20 AgalU were brought to the Department of Pediatrics of Fukuoka University or Kyushu University.
Cases in which GLA activity was found to be <20 AgalU in the mass screening, white blood cell (WBC) GLA activity was measured by a fluorometric enzyme assay. Briefly, whole blood was collected from the neonates and immediately treated with EDTA-2Na. The assay mix included 50 μl leukocyte lysate (WBC pellet prepared from 5 ml blood treated with EDTA-2Na was lysed by sonication in 1.0 ml water) and 50 μl of the reaction mixture (8.0 mM 4-methylumbelliferyl-α-D-galactopyranoside, 100 mM citrate phosphate buffer and 200 mM N-acetylgalactosamine, pH 4.5). This was incubated for 1 h at 37 °C. The reaction was terminated with 1.5 ml of 200 mM glycine buffer (pH 10.7) immediately after the reaction was completed. The fluorescence intensity from 4-MUs was measured with a fluorescent plate reader (Jasco Co, Tokyo, Japan) at 365 and 450 nm.
For genetic analysis, total genomic DNA was extracted from leukocytes of patients. All seven exons of the GLA gene were amplified by PCR, and the amplification products were analyzed by direct sequencing.14 Detailed information on the PCR protocol is available upon request.
Before testing, counseling was provided as to the nature of the disease, its future medical management and risk of recurrence. Informed consent from each child’s parent(s) for newborn mass screening for FD was at birth and that for WBC GLA activity assay and genetic analysis were obtained at their first visit to our hospitals.
All studies were approved by the Ethical Committees of Kumamoto University and Fukuoka University.
Results
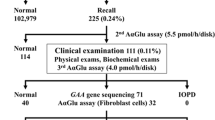
During the 37-month-period from April 2007 to April 2010, a total of 39 224 newborns took the conventional newborn mass screening. Among these, 21 170 (54.0%) newborns were enrolled in this pilot study after written informed consent was provided by the parent(s). The enrollees included 10 827 boys, 10 343 girls. Among the 21 170 newborns, seven (five boys and two girls) showed GLA activity <20 AgalU in the mass-screening test. Six newborns (four boys and two girls) were referred to Fukuoka University Hospital, while one boy (Case 5) was referred to Kyushu University for further examinations. The parents of one girl (Case 7) refused additional medical examinations; therefore, this child was not included in further analyses (Figure 1). All newborns had been delivered without incident at term. Cases 1 and 2 were brothers.

Flow chart of newborn screening for Fabry disease. During a 37-month period from April 2007 to April 2010, 39 224 newborns took the conventional newborn mass screening test in Fukuoka City and its vicinity; this test is given to all newborns in Japan. Among these, 21 170 (54.0%) newborns were enrolled in this pilot study with written informed consent. Seven newborns (five boys and two girls) showed an α-galactosidase A activity <20 AgalU (cutoff value) in the mass screening. The parents of one girl (Case 7) refused additional medical examinations. Four newborns had their white blood cell GLA activities measured, while all six underwent genetic testing. Pathogenic mutations, that is, V199M and IVS4+919G>A, were found in two boys and one boy, respectively. Functional mutations or polymorphisms (see text), that is, E66Q and heterozygous c.−10C>T: g.1170C>T, were found in two boys and one girl, respectively.
One boy (Case 5) presented with neonatal hyperbilirubinemia, high-pitched crying, a dysmorphic face (low-set ears and small mouth), brachydactyly (left shorter fifth finger) and hypotonia. Karyotyping revealed that this boy had a chromosomal abnormality, 46,XY,der(3)t(3;4)(p26;p14). However, we believe that this condition was unrelated to the presence of FD because the GLA gene locus was not involved in his chromosomal abnormality (Table 1).
The mother of one boy (Case 3) had a history of familial renal disease, but the other six subjects had no histories that would suggest a risk of FD. Additional details of the characteristics of familial renal disease in the mother of Case 3 were not available. Except for the dysmorphic features observed in Case 5, none of other six children presented with abnormalities at 1 month of age. The newborns’ parents and other family members with GLA deficiency were counseled and offered medical evaluations and medical follow-up.
WBC GLA activity was very low (3.7 and 4.3 nmol mg−1 protein per hour) in two boys (Cases 1 and 3). In contrast, in one boy and one girl (Cases 4 and 6, respectively), WBC GLA activity was markedly higher, at 31.8 and 20.2 nmol mg−1 protein per hour, respectively, but still well below the normal range of 49.8–116.4 nmol mg−1 protein per hour as determined from 48 healthy volunteers. WBC GLA activity was not measured for two boys (Cases 2 and 5) (Table 1).
Genetic analyses found V199M and IVS4+919G>A mutations in two brothers (Cases 1 and 2) and one boy (Case 3), respectively. V199M is considered pathogenic and is thought to cause the classical phenotype of FD,15 while IVS4+919G>A is also considered pathogenic, but is thought to cause the late-onset phenotype.16, 17 E66Q and heterozygous c.−10C>T: g.1170C>T mutations were found in two boys (Cases 4 and 5) and one girl (Case 6), respectively. These are both functional mutations and are not thought to cause FD, that is, they are considered polymorphisms, but were subjected to further investigations18, 19, 20, 21, 22 (Table 1).
Cases 1–4 were last followed up when the patients were 2–3 years old, by then none of them showed any symptoms of FD.
Discussion
This pilot study of newborn mass screening for FD comprised a large cohort of 21 170 newborns, most of them were of Japanese ethnicity. The screening used a fluorescent GLA activity measurement from a dried blood spot. A total of seven newborns tested positive with respect to low GLA activity (cutoff value: <20 AgalU) and four of these newborns were verified to have low GLA activity by a WBC GLA activity measurement. Thus, the prevalence of positive testing in this screening was 1/3024. Subsequent genetic analysis revealed that six newborns harbored different mutations in the GLA gene. Given that only V199M and IVS4+919G>A are thought to be pathogenic, the prevalence of individuals with pathogenic mutations in our cohort was 1/7057, as three newborns had such mutations. These findings support the contention that the mass-screening system used in this study was legitimate and hence useful for the early diagnosis of FD. At the same time, however, these findings raised concerns about the timing of ERT.
The prevalence of FD in this study (1/7057) suggests that the disease may be far more prevalent than previously thought and much more common than the 1/40 000 males estimated by Desnick et al.1 Nevertheless, this number is lower than those reported in newborn screenings in other countries: 1/1100 boys in Italy (with a 11:1 ratio of late-onset to classic phenotype);23 1/3859 boys and girls in Austria (no instances of classic phenotype, but one case in which the phenotype could not be determined)24 and 1/1250 boys in Taiwan (86% with the late-onset mutation IVS4+919G>A).17 These differences may have to do with the genetic backgrounds of the newborns examined in each study.
GLA activities measured by the fluorescence assay were in accordance with GLA activities measured from WBC samples, an accepted method for GLA measurement in the diagnosis of FD. This again supports the notion that our screening system, which utilizes a fluorescence assay to measure GLA activity, is reliable. WBC GLA activities were very low, that is, 3.7 and 4.3 nmol mg−1 protein per hour, in Cases 1 and 3, respectively. The mutations identified in these newborns were V199M and IVS4+919G>A in the GLA gene, which are considered pathogenic mutations and are thought to cause classical and late-onset phenotypes of FD, respectively. In contrast, WBC GLA activity was markedly higher, at 31.8 and 20.2 nmol mg−1 protein per hour, in Cases 4 and 6, respectively, but still well below the normal range of 49.8–116.4 nmol mg−1 protein per hour. The mutations identified in these two cases are functional mutations and are considered polymorphisms that lead to low GLA activity but may not evolve into FD; however, whether they are totally benign polymorphisms is still controversial.18, 19, 20, 21, 22 Overall, the GLA activity measurements in this study seemed to reflect the nature of the identified GLA gene mutations.
Given that newborn mass screening for FD is conducted to allow for early diagnosis and, in turn, early treatment with ERT, this early diagnosis raises a number of concerns. First, the timing of ERT is still controversial, and there is uncertainty as to when ERT should be initiated in neonates (many of whom will be asymptomatic). Several studies have demonstrated that ERT must be administered before instances of renal or cardiac failure in order to achieve optimal results.10, 11, 12 However, Ross25 insists that ‘premature treatment may cause more harm than good’ as a result of ‘side-effects’ and ‘medicalizing of a normal childhood.’ Others, instead, indicate that there is no evidence that early ERT is ineffective or that it harms the patient.26, 27, 28, 29, 30 Indeed, the findings that are currently available on the efficacy and side effects of ERT are based on a small sampling and are the result of very few ERT follow-up reports for young children. Therefore, the implementation of mass screening for FD, which has only recently been initiated in very limited regions of the world, should provide significant insights into these controversies. In addition, it will be necessary to accumulate enough long-term follow-up data on the efficacy and safety of ERT to conclusively determine the effectiveness of ERT in newborns diagnosed with FD during newborn mass screening; as the prevalence of FD is very low, this process may take many years.
Second, in relation to the first concern raised above, neither GLA activity nor genotypes necessarily correlate with phenotypes in terms of severity and onset of the disease. This is particularly the case in heterozygous females who may have significantly reduced enzyme activity but no symptoms. Of course, positive results in newborn screening followed by further examinations, including WBC GLA measurement and genetic analyses, enable ERT to be initiated at first onset of the condition and alert the child’s female relatives to the necessity of future medical examinations. Serum or urinary globotriaosylceramide or serum globotriaosyl spingosine levels may give us better clues for anticipating the severity and onset of the disease identified in newborn mass screening, although they were not investigated in the present study.
Third, there are also ethical issues associated with early screening for FD. Newborns who tested positive in newborn mass screening may be given a presymptomatic diagnosis by genetic testing. According to the Japanese Association of Medical Science, presymptomatic diagnosis by genetic testing should only be performed after the examinee has sufficiently understood the available preventive measures and therapeutic strategies.31 Obviously, imparting such information to a neonate is impossible, but providing a parent with sufficient counseling to making an informed decision is essential. Thus, careful genetic counseling is required, preferentially from a knowledgeable geneticist.
Likewise, adequate education on the symptoms and disease progression for FD should also be provided to parents and their relatives. On their first visit to our hospitals, parents were unfamiliar with FD, and, to ease their concerns, it was necessary to provide them detailed information about its nature, future medical management and risk of recurrence. Furthermore, as FD is an X-linked inherited disease, inadequate counseling may create a burden to mothers and even affect family relationships, which may lead to serious consequences, such as divorce. From our experience with the seven cases reported in this study, some parents, for whatever reason, were surprised that FD might be serious, while others were overly alarmed by the disease.
To overcome all concerns raised, certain guidelines for management of FD, including when ERT is appropriate, should be established based on evidence collected as a result of accumulating experiences with FD identified in newborn mass screening. Although FD is quite rare, it is feasible to establish such guidelines, as guidelines are available for similar rare diseases, that is, Gaucher disease32 and Pompe disease.33 These worldwide guidelines have proven to be extremely useful.32, 33 In addition, these guidelines could aid in the collection of global data that would assist in determining not only the appropriate age for ERT, but also the nature and duration of follow-up care and genetic counseling, providing, in short, a means of coordinating both research and treatment.
References
Desnick, R. J., Ioannou, Y. A. & Eng, C. M. Alpha-galactosidase A Deficiency: Fabry Disease, in The Metabolic and Molecular Bases of Inherited Disease 8th edn eds Scriver C. R., Beaudet A. L., Sly W. S., Valle D., 3733–3774 (McGraw-Hill: New York, NY, 2001).
Mehta, A., Beck, M., Eyskens, F., Feliciani, C., Kantola, I., Ramaswami, U. et al. Fabry disease: a review of current management strategies. QJM. 103, 641–659 (2010).
Nakamura, K., Hattori, K. & Endo, F. Newborn screening for lysosomal storage disorders. Am. J. Med. Genet. C. Semin. Med. Genet. 157, 63–71 (2011).
Ramaswami, U., Whybra, C., Parini, R., Pintos-Morell, G., Mehta, A., Sunder-Plassmann, G. et al. Clinical manifestations of Fabry disease in children: data from the Fabry Outcome Survey. Acta. Paediatr. 95, 86–92 (2006).
Eng, C. M., Fletcher, J., Wilcox, W. R., Waldek, S., Scott, C. R., Sillence, D. O. et al. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J. Inherit. Metab. Dis. 30, 184–192 (2007).
Matsuzawa, F., Aikawa, S., Doi, H., Okumiya, T. & Sakuraba, H. Fabry disease: correlation between structural changes in alpha-galactosidase, and clinical and biochemical phenotypes. Hum. Genet. 117, 317–328 (2005).
Eng, C. M., Guffon, N., Wilcox, W. R., Germain, D. P., Lee, P., Waldek, S. et al. Safety and efficacy of recombinant human alpha-galactosidase A—replacement therapy in Fabry’s disease. N. Engl. J. Med. 345, 9–16 (2001).
Schiffmann, R., Kopp, J. B., Austin, H. A. 3rd, Sabnis, S., Moore, D. F., Weibel, T. et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 285, 2743–2749 (2001).
Beck, M., Ricci, R., Widmer, U., Dehout, F., de Lorenzo, A. G., Kampmann, C. et al. Fabry disease: overall effects of agalsidase alfa treatment. Eur. J. Clin. Invest. 34, 838–844 (2004).
Wilcox, W. R., Banikazemi, M., Guffon, N., Waldek, S., Lee, P., Linthorst, G. E. et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am. J. Hum. Genet. 75, 65–74 (2004).
Banikazemi, M., Bultas, J., Waldek, S., Wilcox, W. R., Whitley, C. B., McDonald, M. et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann. Intern. Med. 146, 77–86 (2007).
Weidemann, F., Breunig, F., Beer, M., Sandstede, J., Turschner, O., Voelker, W. et al. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation 108, 1299–1301 (2003).
Chamoles, N. A., Blanco, M. & Gaggioli, D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin. Chim. Acta. 308, 195–196 (2001).
Nakamura, K., Sekijima, Y., Nakamura, K., Hattori, K., Nagamatsu, K., Shimizu, Y. et al. Cerebral hemorrhage in Fabry's disease. J. Hum. Genet. 55, 259–261 (2010).
Shabbeer, J., Yasuda, M., Luca, E. & Desnick, R. J. Fabry disease: 45 novel mutations in the alpha-galactosidase A gene causing the classical phenotype. Mol. Genet. Metab. 76, 23–30 (2002).
Ishii, S., Nakao, S., Minamikawa-Tachino, R., Desnick, R. J. & Fan, J. Q. Alternative splicing in the alpha-galactosidase A gene: increased exon inclusion results in the Fabry cardiac phenotype. Am. J. Hum. Genet. 70, 994–1002 (2002).
Hwu, W. L., Chien, Y. H., Lee, N. C., Chiang, S. C., Dobrovolny, R., Huang, A. C. et al. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the lateronset GLA mutation c.936+919G4A (IVS4+919G4A). Hum. Mutat. 30, 1397–1405 (2009).
Lee, B. H., Heo, S. H., Kim, G.-W., Park, J.-Y., Kim, W.-S., Kang, D. H. et al. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J. Hum. Genet. 55, 512–517 (2010).
Togawa, T., Tsukimura, T., Kodama, T., Tanaka, T., Kawashima, I., Saito, S. et al. Fabry disease: Biochemical, pathological and structural studies of the α-galactosidase A with E66Q amino acid substitution. Mol. Genet. Metab. 105, 615–620 (2012).
Davies, J. P., Winchester, B. G. & Malcolm, S. Sequence variations in the first exon of alpha-galactosidase A. J. Med. Genet. 30, 658–663 (1993).
Oliveira, J. P., Ferreira, S., Reguenga, C., Carvalho, F. & Månsson, J. E. The g.1170C>T polymorphism of the 5′ untranslated region of the human alpha-galactosidase gene is associated with decreased enzyme expression—evidence from a family study. J. Inherit. Metab. Dis. 31, S405–S413 (2008).
Oliveira, J. P., Ferreira, S., Barceló, J., Gaspar, P., Carvalho, F., Sá Miranda, M. C. et al. Effect of single-nucleotide polymorphisms of the 5′ untranslated region of the human α-galactosidase gene on enzyme activity, and their frequencies in Portuguese caucasians. J. Inherit. Metab. Dis. 31, S247–S253 (2008).
Spada, M., Pagliardini, S., Yasuda, M., Tukel, T., Thiagarajan, G., Sakuraba, H. et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am. J. Hum. Genet. 79, 31–40 (2006).
Mechtler, T. P., Stary, S., Metz, T. F., De Jesús, V. R., Greber-Platzer, S., Pollak, A. et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet 379, 335–341 (2012).
Ross, L. F. Newborn screening for lysosomal storage diseases: an ethical and policy analysis. J. Inherit. Metab. Dis. 35, 627–634 (2012).
Ries, M., Clarke, J. T., Whybra, C., Timmons, M., Robinson, C., Schlaggar, B. L. et al. Enzyme-replacement therapy with agalsidase alfa in children with Fabry disease. Pediatrics 118, 924–932 (2006).
Ries, M., Clarke, J. T., Whybra, C., Mehta, A., Loveday, K. S., Brady, R. O. et al. Enzyme replacement in Fabry disease: pharmacokinetics and pharmacodynamics of agalsidase alpha in children and adolescents. J. Clin. Pharmacol. 47, 1222–1230 (2007).
Schiffmann, R., Martin, R. A., Reimschisel, T., Johnson, K., Castaneda, V., Lien, Y. H. et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J. Pediatr. 156, 832–837 (2010).
Ramaswami, U., Wendt, S., Pintos-Morell, G., Parini, R., Whybra, C., Leon Leal, J. A. et al. Enzyme replacement therapy with agalsidase alfa in children with Fabry disease. Acta. Paediatr. 96, 122–127 (2007).
Ramaswami, U. Update on role of agalsidase alfa in management of Fabry disease. Drug Des. Devel. Ther. 5, 155–173 (2011).
The Japanese Association of Medical Sciences “Guidelines for Genetic Tests and Diagnoses in Medical Practice” http://jams.med.or.jp/guideline/genetics-diagnosis_e.pdf.
Martins, A. M., Valadares, E. R., Porta, G., Coelho, J., Semionato Filho, J., Pianovski, M. A. et al. Recommendations on diagnosis, treatment, and monitoring for Gaucher disease. J. Pediatr. 155, S10–S18 (2009).
Cupler, E. J., Berger, K. I., Leshner, R. T., Wolfe, G. I., Han, J. J., Barohn, R. J. et al. Consensus treatment recommendations for late-onset Pompe disease. Muscle Nerve 45, 319–333 (2012).
Acknowledgements
This work was supported in part by a Grant-in-Aid for Scientific Research (A) (#21249062 to SH), for Challenging Exploratory Research (#23659529 and 25670481 to SH), for Scientific Research on Innovative Areas (25129708 to SH) for Bilateral Joint Research Projects (SH) from Japan Society for the Promotion of Science (JSPS); a Grant-in-Aid for Scientific Research on Innovative Areas ‘Genome Science’ (SH) from the Ministry of Education, Culture, Sports, Science and Technology; Research Grants (21B-5 to SH) for Nervous and Mental Disorder, and Health and Labour Science Research Grant (21210301 and KB220001 to SH) and Grant-in-Aid for the Research on Measures for Intractable Diseases (No. H22-Nanji-Ippan-49 to SH) from the Ministry of Health, Labour and Welfare; the Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University (SH); Research grants for Central Research Institute for the Molecular Pathomechanisms of Epilepsy of Fukuoka University (SH) and Recommended Projects from Fukuoka University (#117016 to SH); Adaptable and Seamless Technology Transfer Program through Target-driven R&D (A-STEP) Exploratory Research, Japan Science and Technology Agency; and Research Grant from the Japan Epilepsy Research Foundation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Inoue, T., Hattori, K., Ihara, K. et al. Newborn screening for Fabry disease in Japan: prevalence and genotypes of Fabry disease in a pilot study. J Hum Genet 58, 548–552 (2013). https://doi.org/10.1038/jhg.2013.48
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.48
Keywords
This article is cited by
-
Clinical and neuroimaging review of monogenic cerebral small vessel disease from the prenatal to adolescent developmental stage
Japanese Journal of Radiology (2024)
-
A nationwide cross-sectional analysis of biopsy-proven Fabry nephropathy: the Japan Renal Biopsy Registry
Clinical and Experimental Nephrology (2023)
-
Development and validation of a disease-specific quality of life scale for adult patients with Fabry disease in Japan
Journal of Patient-Reported Outcomes (2022)
-
Fabry disease associated with multiple myeloma: a case report
CEN Case Reports (2022)
-
DNA methylation impact on Fabry disease
Clinical Epigenetics (2021)
