
Abstract
Local genomic architecture, such as segmental duplications (SDs), can induce copy number variations (CNVs) hotspots in the human genome, many of which manifest as genomic disorders. Significant technological advances have been achieved for genome-wide CNV investigations, but these costly methods are not suitable for genotyping certain disease-associated CNVs or other loci of interest in populations. Recently, two independent studies showed that the murine meiosis expressed gene 1 (Meig1) was critical to spermatogenesis. We found that the human orthologue MEIG1 is flanked by an SD pair, between which non-allelic homologous recombination (NAHR) can cause recurrent CNVs. To study this potential CNV hotspot and its role in spermatogenesis, we developed a new CNV genotyping method, AccuCopy, based on multiplex competitive amplification to investigate 320 patients with spermatogenic impairment and 93 healthy controls. Three MEIG1 duplications (two in patients and one in controls) were identified, whereas no deletion was found. As NAHR results in more recurrent deletions than duplications at a locus, the over representation of recurrent MEIG1 duplications suggests a potential purifying selection operating on this hotspot, possibly via fecundity. We also showed that AccuCopy is an efficient and reliable method for multiplex CNV genotyping.
Similar content being viewed by others


Introduction
Copy number variation (CNV), which is generated by genomic rearrangement in the human genome, has been recognized as one of the main genetic factors underlying genomic disorders and other human diseases.1 Accurate and efficient CNV genotyping is one of the key steps when studying CNV-associated human diseases.
Various molecular mechanisms have been elucidated for CNV mutations, including non-allelic homologous recombination (NAHR), a DNA recombination-based mechanism that takes long DNA repeats as substrates for homologous recombination and causes CNVs of the genomic region flanked by repeats.2 Notably, the NAHR mechanism has been found to have an important role in causing CNV hotspots in the human genome.3, 4, 5 In this case, the main recombination substrates are segmental duplications (SDs) or low-copy repeats, which are repeated genomic segments of >1 kb in length and showing >90% sequence similarity. SDs account for ∼5% of the human genome.2, 6
Male infertility caused by Y chromosomal microdeletions is a typical genomic disorder, the classical types of which are recurrent deletions caused by NAHR between repeat sequences.7, 8, 9, 10 Whereas frequent recurrent deletions in the human Y chromosome have been robustly associated with spermatogenic impairment, the role of autosomal CNVs and their affected genes in spermatogenesis has not been examined systematically.
Recently, two studies utilizing knockout mouse models reported that the murine meiosis expressed gene 1 (Meig1) was critical in spermatogenesis.11, 12 Meig1 is a murine gene with testis-specific expression. Zhang et al.11 found that homozygous Meig1-knockout male mice are viable but sterile, whereas no obvious defect was detected in heterozygous Meig1-knockout mice. The stage of elongation and condensation of spermatogenesis was shown to be significantly impaired.11 Salzberg et al.12 obtained similar results showing that developing germ cells rarely differentiated past the spermatogenesis stage in their Meig1−/− male mice; the few sperms that managed to reach the epididymis were totally immotile. These consistent findings indicated that the murine Meig1 gene was crucial for spermatogenesis in mice.
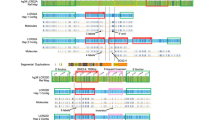
MEIG1, the human orthologue of Meig1, maps to chromosome 10p13 of the human genome (Figure 1). Interestingly, the MEIG1 gene is flanked by two direct SDs (SD1 and SD2), which can function as substrates for NAHR and consequently mediate recurrent deletions and duplications of MEIG1 gene (Figure 1). We hypothesized that the SD-mediated NAHR events may be frequent and the locus surrounding MEIG1 is a potential CNV hotspot.

Local genomic architecture surrounding the MEIG1 gene and identification of three recurrent MEIG1 duplications. Above, the genomic architecture of MEIG1 and its flanking region in UCSC Genome Browser (hg19). The MEIG1 gene is flanked by a pair of homologous SDs in direct orientation (SD1 and SD2). There is a complex genomic region with some other repeat sequences between SD1 and SD2. Recurrent MEIG1 duplications and deletions are hypothesized to be generated between SD1 and SD2. Therefore, four target genomic segments (S1–S4) were selected for the AccuCopy assay to investigate the hypothesized CNVs. The positions of S1–S4 were indicated by arrows, and the positions of SD1 and SD2 were shown by gray shadow in the aCGH data plot (below). High-resolution oligonucleotide aCGH analyses confirmed all three MEIG1 duplications identified by AccuCopy in subjects C13, P16 and P65. The green (reduction), black (no obvious change), and red (increase) dots show the relative intensities of tested genomic DNAs compared to a reference DNA in log2 ratio (deviation from horizontal line of 0) and genomic locations of the oligonucleotide probes used in our aCGH assay. The regions that lack unique probes correspond to repeat sequences such as SDs. The duplicated genomic segments are indicated with red horizontal bars. The breakpoints of these recurrent duplications are located within homologous SD1 and SD2.
To study the potential CNV hotspot of MEIG1 and its role in spermatogenesis, we developed a multiplex CNV genotyping method, named ‘AccuCopy’, to investigate the MEIG1 CNVs in 320 Chinese patients with spermatogenic impairment and 93 healthy male controls. The AccuCopy method is based on competitive PCR amplification (Figure 2) and can interrogate CNV status at multiple genomic loci in the same assay reaction. The MEIG1 CNVs identified by AccuCopy can be subsequently confirmed by high-resolution comparative genomic hybridization (CGH) microarray and short tandem repeat (STR) genotyping.

Illustration of the AccuCopy method for multiplex copy number quantification. The example of one reference locus (R0) and four target genomic segments (T1–T4) with various copy numbers of 1–4 was illustrated. Generally, single-copy gene (that is, two copies in a diploid genome) is selected as reference. Long competitive double-strand DNA sequence is synthesized for each target and reference genomic segment. Each competitive DNA sequence (C) is highly similar to its human homology in sample DNAs (S), while they are different in length for several base pairs (bp). The red bars in this figure indicate the oligonucleotide sequences in human homologies that are deleted in their competitive DNA sequences. The black bar indicates the 5′ flanking sequence of the deleted part and the gray bar indicates its 3′ flanking sequence. Both of these flanking sequences are identical between human homology and its competitive DNA. The synthesized competitive DNAs for target and reference segments are first mixed with a defined amount of genomic DNA of tested sample. Multiplex fluorescence competitive PCR is performed to simultaneously amplify all reference and target segments from both sample DNA and competitive DNA. The blue dots indicate a blue fluorescent group labeled to each forward primer. Amplicons with different sizes are separated by capillary electrophoresis and the peak ratio of sample DNA to competitive DNA (S/C) for each segment is calculated. After normalization by reference segment's peak ratio, the copy number ratio of each target segment to reference can be easily determined by its peak ratios divided by reference's peak ratio, when the competitive DNA segments are well balanced in molecular number in the DNA mixture. The copy number of target segment can be identified, given that the copy number of reference segment is known.
Materials and methods
Subjects
Study subjects were volunteers from the affiliated hospitals of Nanjing Medical University between July 2007 and July 2010 (NJMU Infertile Study).13 The protocol and consent form were approved by the Institutional Review Boards of Nanjing Medical University and School of Life Sciences in Fudan University before the study. All activities involving human subjects were done under full compliance with government policies and the Helsinki Declaration.
The patients were diagnosed with unexplained male infertility. Men with abnormal sexual and ejaculatory functions, immune infertility, semen non-liquefaction, medical history of risk factors for infertility and receiving treatment for infertility were excluded from the study. In order to avoid azoospermia or severe oligozoospermia frequently caused by Y chromosome microdeletions, we excluded subjects with Y chromosome microdeletions of azoospermia factor regions (AZFa, AZFb and AZFc).8, 9, 10 The semen analysis for sperm concentration, motility and morphology was performed following the World Health Organization criteria.14 In this study, the patients with spermatogenic impairment included both azoospermia (no sperms in the ejaculate even after centrifugation) and oligozoospermia (sperm counts <20 × 106 ml−1).
The controls were healthy and fertile young men who had fathered one or more healthy children without assisted reproductive measures from the same hospital during the same period. The final analyses included 320 eligible patients and 93 eligible controls. All subjects were genetically unrelated ethnic Han-Chinese from East China.
CNV genotyping using the AccuCopy method
We newly developed the AccuCopy method to accurately interrogate CNV status at multiple genomic loci. The basic molecular principle of competitive PCR amplification for AccuCopy is illustrated in Figure 2.
To investigate the hypothesized recurrent MEIG1 CNVs mediated by NAHR between highly similar SD1 and SD2 (Figure 1), we chose four target genomic segments within the CNV region for the AccuCopy assay, including S1 within the DCLRE1C gene, S2 in 3′-untranslated region of the MEIG1 gene, S3 adjacent to 5′-untranslated region of MEIG1 and S4 in exon 1 of MEIG1 (Figure 1). The reference genome sequences were obtained from the UCSC Genome Browser (hg19; http://genome.ucsc.edu). The forward and reverse primers of S1–S4 were provided in Supplementary Table 1.
The size of PCR product amplified from human genomic DNA is 179, 126, 202 and 152 bp for S1, S2, S3 and S4 primer pairs, respectively.
In addition to these primers for target segments, four reference segments that were utilized for normalization, were screened and chosen at four loci of POLR2A, POP1, RPP14 and TBX15. Their primers are provided in Supplementary Table 1.
The size of PCR product amplified from human genomic DNA is 115, 99, 144 and 187 bp for POLR2A, POP1, RPP14 and TBX15 primer pairs, respectively. The primers for both reference and target segments were synthesized and the forward primers were fluorescent-labeled at Sangon Biotech (Shanghai, China).
The competitive DNAs for the four reference and four target segments were synthesized in double strand and provided in a mixture from Genesky Biotechnologies (Shanghai, China). These competitive DNAs are almost same as their homologies in the human reference genome except two base pairs deleted. Their sequences are listed in Supplementary Figure 1. The synthesized competitive DNAs for target and reference segments are first mixed with a defined amount of genomic DNA of tested sample, and then subject to a multiplex fluorescence competitive PCR amplification that can simultaneously amplify all reference and target segments from both the sample DNA and competitive DNA using multiple fluorescence-labeled primer pairs. The PCR product of competitive DNAs is 2 bp shorter than that of human genomic DNAs amplified by the same pair of primers; therefore can be distinguished from each other after fluorescence capillary electrophoresis.
The PCR reaction was prepared in 20 μl for each sample, containing 1 × Multiplex PCR Master Mix (Qiagen, Valencia, CA, USA), 0.05 μM each primer, 1 × Competitive DNA Mix (Genesky Biotechnologies) and ∼10 ng genomic DNA. The PCR program was as follows: 95 °C 10 min; 11 cycles of 94 °C 20 s, 65−0.5 °C/cycle 40 s, 72 °C 1.5 min; 24 cycles of 94 °C 20 s, 59 °C 30 s, 72 °C 1.5 min; and 60 °C 60 min.
PCR products were diluted 20-fold before being run by capillary electrophoresis using ABI (Carlsbad, CA, USA) 3730XL genetic analyzer. Raw data were analyzed by GeneMapper 4.0 (ABI) and the height and area data for all specific peaks were exported into a Microsoft Excel file. The sample/competitive (S/C) peak ratio was calculated for all four target segments and four reference segments. The S/C ratio for each target fragment was first normalized based on four reference segments, respectively. The four normalized S/C ratios were further normalized to the median value in all samples for each reference segment, respectively, and then averaged. If one of the four normalized S/C ratios deviated >25% from the average of the other two, it was excluded for further analysis. The copy number of each target segment was determined by the average S/C ratio times two, given that the copy numbers of four reference segments are two in the diploid genome.
DNA sequencing of PCR amplification products and STR genotyping
Any copy number loss suggested by AccuCopy should be verified by DNA sequencing to exclude possible DNA variations in the primer regions. In addition, a GA di-nucleotide STR marker within the MEIG1 gene (located between SNPs rs71910374 and rs6602772) was genotyped by DNA sequencing to examine whether the identified CNVs with the same size share a common mutation event or alternatively be recurrent.
DNA sequencing is performed using the Sanger dideoxy method on an ABI 3730XL genetic analyzer. DNA sequences were analyzed by comparing to the human genome reference assembly (hg19) with the BLAT tool of the UCSC Genome Browser (http://genome.ucsc.edu/cgi-bin/hgBlat?command=start).
Array-based CGH (aCGH)
The oligonucleotide-based CGH microarray is an efficient and reliable assay for CNV studies.15 In this study, we designed a high-resolution aCGH assay based on the Agilent 8 × 60K format to finely examine the hypothesized recurrent MEIG1 CNVs and other loci in the human genome. The aCGH resolution for the MEIG1 CNV region is <500 bp between oligonucleotide probes. Investigating probes were selected from the Agilent eArray system (http://earray.chem.agilent.com/earray/). Probes having sequences complementary to more than one genomic locus have been purged and only unique sequence probes were used.
Subjects C13, P16 and P65, which were suggested to have copy number gains consistently across all four target segments (S1–S4) as candidates for the hypothesized recurrent MEIG1 duplications, were further analyzed by oligonucleotide-based aCGH. DNA processing, microarray handling and data analysis were conducted by following the Agilent oligonucleotide aCGH protocol (version 5.0). The Agilent Genomic Workbench software (v6.5.0.18) was used for calling CNVs, while ADM-2 was chosen as the analysis algorithm.
Results
CNVs of the MEIG1 gene identified by the AccuCopy method
To investigate the hypothesized recurrent MEIG1 CNVs (Figure 1), we used the AccuCopy assay. The MEIG1 assay of AccuCopy in 320 patients with spermatogenic impairment (P1–P320) and 93 normal controls (C1–C93) was summarized in Figure 3. Most of the subjects were shown to have two copies consistently across the four target segments, suggesting a neutral CNV status in a diploid genome. Obviously increased or reduced amplification of one or all of four target segments were indicated by AccuCopy in five subjects. Increased copy numbers were identified across S1–S4 segments in subjects C13 (S1=3.00±0.02, S2=3.26±0.02, S3=3.18±0.03 and S4=3.10±0.02), P16 (S1=3.37±0.05, S2=3.30±0.05, S3=3.23±0.04 and S4=3.22±0.05) and P65 (S1=3.39±0.08, S2=3.24±0.08, S3=3.35±0.08 and S4=3.28±0.08), which were consistent with the hypothesized recurrent duplications of MEIG1. Subjects P139 (S2=0.99±0.02) and P274 (S2=0.97±0.01) were found to be reduced in the amplification of S2, while the copy numbers in the remaining three segments were neutral. The plot of capillary electrophoresis of the AccuCopy assay, showing three examples with neutral copy number, loss and gain, respectively, were illustrated in Supplementary Figure 2.

Assays of the CNVs in the MEIG1 locus and its flanking regions using the AccuCopy method. The copy number states of four target DNA segments (S1–S4) for each patient (P) or control (C) were shown in four panels. (a) S1; (b) S2; (c) S3; and (d) S4. Each column indicates a patient or control. The columns for 93 controls are shadowed. The copy number states for each target segment were measured four times by comparing with four reference segments. The mean values were shown in column height; whereas the standard deviations were indicated by the bar on top of each column. The neutral copy numbers for all these four autosomal segments are two. The AccuCopy assay showed copy number gains (three copies) in subjects C13, P16 and P65, consistently across all the four target segments. Two possible copy number losses (one copy; they were confirmed to be false positive by further assays) were only suggested for the S2 segment in subject P139 and 274.
Reduced amplification potentially caused by mismatched primer
The reduced amplification of only S2 in subjects P139 and P274 may suggest a small exonic deletion of MEIG1. However, an alternative explanation for this phenomenon is mismatching between S2 primers and the genomic DNAs of subjects P139 and P274 resulting in allele-specific amplification. As AccuCopy is based on competitive PCR amplification, even a mismatch of one base pair in the long primer sequence can potentially cause reduced amplification, which could be misinterpreted as a copy number loss. A possible mismatch in primer regions can be identified by DNA sequencing.
Therefore, the S2 amplification primer annealing regions were amplified and sequenced in subjects P139 and P274. Using the BLAT tool of UCSC Genome Browser, we identified a heterozygous C–T variation at the 18th nucleotide of the 29-mer S2R primer annealing regions in both P139 and P274, but none in copy-number-neutral subjects (Supplementary Figure 3). This single-nucleotide variation, which located in the 3′-untranslated region of MEIG1 gene, has not been included in the NCBI dbSNP database, and it can cause mismatching between primer S2R and the sample DNAs of P139 and P274, and may consequently results in the reduced amplification of S2 in these two subjects (Figure 3). Therefore, the DNA sequence analysis suggested that the reduced amplification of S2 in subjects P139 and P274 was due to primer mismatching rather than copy number loss of S2.
Recurrent MEIG1 duplications confirmed by aCGH and STR genotyping
Different from the reduced amplification of P139 and P274 only in S2 segment, the AccuCopy assay showed consist increased amplification across S1–S4 segments in C13, P16 and P65 (Figure 3), suggesting recurrent MEIG1 duplications mediated by NAHR between SD1 and SD2 (Figure 1). All three duplications suggested by AccuCopy were confirmed to be ∼82 kb in length by aCGH (Figure 1). Two out of the three duplications were found in 320 patients and one in 93 controls, and no significant differences was suggested between these two subject groups (P=0.54, Fisher's exact test). On the basis of these observations, MEIG1 duplications may be neutral on male fecundity.
In addition, our aCGH assay had a very high resolution and revealed that both proximal and distal ends of three MEIG1 duplications were located in SD1/SD2, consistent with the hypothesized recurrent MEIG1 duplications. The aCGH assay also showed that these three recurrent duplications affected the whole MEIG1 gene and the 5′ region of DCLRE1C as well, a gene involved in V(D)J recombination and human combined immune deficiency.16, 17
A GA tandem repeat marker within the MEIG1 gene was genotyped by DNA sequencing to test whether three duplications with the same size share a common mutation event or recurrently resulted from independent events. The STR genotypes are as follows: C13, 13/20/28; P16, 12/20/26; and P65, 12/12/13. No common duplication allele (that is, sharing the same repeat numbers in two repeat positions) was identified, even if a one-step STR slippage mutation is allowed after a hypothesized common ancestral duplication event. Therefore, a common ancestor shared by these three duplications is not readily supported. An alternative explanation of recurrent MEIG1 duplications is preferred.
Discussion
AccuCopy is an efficient and reliable assay for multiple CNV genotyping
Some advanced technologies, including oligonucleotide CGH microarrays, SNP genotyping microarrays, and some assays based on next-generation sequencing, have been frequently utilized in genome-wide studies on CNVs.1 However, most of these genome-wide CNV assays are costly, and may not be the first choice for the CNV studies on a specific gene or only a few genetic loci of interest. Therefore, efficient and reliable CNV assays for target regions are important tools for investigating the roles of CNVs in human diseases and other traits. Quantitative PCR assay was a popular method for gene expression studies and can also be alternatively used for measurement of gene copy numbers; however, our previous study showed that the CNV resolving power of quantitative PCR is not satisfying and three or more repeated amplifications for the same assay is required to genotype a CNV.18 Armour et al. developed an accurate method known as the paralogue ratio test, which could be used for rapid and high-throughput CNV genotyping for a specific locus.19 However, the paralogue ratio test can only be used to interrogate regions containing paralogous sequences in the same genome and cannot be extended to other unique genomic regions.
Multiplex genotyping is also an additional characteristic needed for CNV analyses. In this study, we introduced the new method of AccuCopy for multiple CNV genotyping. AccuCopy has been verified to be able to amplify and measure at least 12 loci in the same amplification reaction. In addition to the four reference segments and four target segments from the MEIG1 region, four more segments outside MEIG1 were also included in the same assay (data not shown). The AccuCopy assay of up to 18 loci was also verified in our lab.
Accurate copy number measurements can be achieved by normalization of copy numbers in tested segments to those in reference segments with known or neutral copy numbers (Figure 2). When we investigated the loci that we were interested in, one or more reference segments were chosen from the regions free of CNVs based on the DGV database. Therefore, the effects of known CNVs in human populations on data normalization can be avoided. However, other frequent CNVs that have not been archived in DGV and rare CNVs still exist and can affect CNV genotyping. Accordingly, we included four reference segments in this study, including POLR2A, POP1, RPP14 and TBX15. In this study, all the CNV values normalized by POLR2A, POP1, RPP14 and TBX15 were consistent, suggesting these four reference segments were stable and good candidate regions for reference in the AccuCopy assay.
As the AccuCopy technology is based on competitive amplification, the possible mismatching between primers designed according to the human reference genome and the test DNAs with unknown SNPs and/or other rare mutations can reduce PCR amplification markedly and consequently lead to false-positive copy number loss. Such examples have been found for the target segment S2 in subjects P139 and P274 (Figure 3). Therefore, DNA sequencing of primer regions is highly recommended to verify any copy umber loss suggested by AccuCopy (Supplementary Figure 3). Besides, multiple segments for each target locus were suggested to be included for accurate CNV genotyping. Actually, the multiplex capability of the AccuCopy technology can address this issue very well.
In this study, three recurrent MEIG1 duplications were identified consistently across four target segments and were confirmed by high-resolution CGH microarrays, demonstrating that AccuCopy was a reliable assay for multiple CNV genotyping. Although currently the multiplex ligation-dependent probe amplification is a major technology for multiple CNV genotyping and has been widely used for the studies on genomic disorders and genomic imbalances,20, 21 the newly developed AccuCopy method displays a promising advantage on efficiency (<2 h per assay), simple operation and also a limited amount of sample DNA required (only 10 ng or less per assay).
SD-mediated CNV hotspots in the human genome
Repeated sequences (for example, SDs, SINEs and LINEs) are prevalent in the human genome.22, 23 The NAHR mechanism between homologous repeat pairs is one of the important mutational mechanisms for CNVs.2, 24, 25 Notably, our previous studies showed that the CNVs mediated by NAHR between SDs rank higher than all the others based on mutation rate.4 The SD-mediated CNVs have been regarded as mutational hotspots in the human genome.3
It has been hypothesized that the mutation rate of SD-mediated recurrent CNVs is associated with SD length, distance and similarity between SD pairs.18 Intriguingly, the genetic observations in the microdeletions in AZFc, a Y chromosomal region with various palindrome repeats, revealed that the deletions mediated by long repeats have higher mutation rate than other deletions in the same region.13 Similar findings have recently been obtained in the region associated with Smith–Magenis microdeletion syndrome and Potocki-Lupski microduplication syndrome.5 Therefore, the NAHR mechanism and the distribution of long SD pairs can help us predict unknown CNV hotspots in the human genome.
Our studied MEIG1 region is flanked by two SDs with high sequence similarity of 95% in direct orientation (Figure 1). In addition, these two SDs are only 82 kb apart from each other, much shorter than some known CNV hotspots associated with genomic disorders (from hundreds of kb to several Mb). Therefore, frequent NAHR events between these SDs are hypothesized and the MEIG1 is potentially a CNV hotspot. In the DGV database of the latest version with 66 741 CNV records (2 November 2010), only one deletion (variation 23 238) identified by Korbel et al.26 via paired-end mapping perfectly matched recurrent MEIG1 CNVs and have both breakpoints located in SD1 and SD2 flanking MEIG1. Notably, some DGV-recorded CNVs are overlapped with recurrent MEIG1 CNVs and can be regarded as candidates for recurrent CNVs, as the majority of previous genome-wide CNV genotyping methods do not have enough resolution to finely map the CNV breakpoints within SD1/SD2 of only 12 kb in length. In this study, we identified two recurrent MEIG1 duplications in 320 patients with spermatogenic impairment (0.6%) and one in 93 healthy controls (1.1%). Taking the aforementioned observations together, it was suggested that the MEIG1 region was a CNV hotspot mediated by local SD architecture.
Over representation of duplication and purifying selection on recurrent MEIG1 CNVs
Size, location and deletion/duplication ratio of the recurrent CNVs generated by the NAHR mechanism are predictable, which is different from the CNVs caused by other molecular mechanisms. NAHR can mediate more recurrent deletions than duplications in any locus flanked by SD pairs.2 Notably, the experimental observations supported this theoretical prediction. Turner et al.27 studied three autosomal and one Y chromosomal NAHR-mediated CNVs using pooled sperm typing and found that the ratios of deletion to duplication were ∼2:1 for autosomal loci and 4:1 for human Y chromosomal loci. However, we only identified three recurrent MEIG1 duplications in 413 subjects but none for deletion in this study. Given the deletion/duplication ratio of 2:1 for autosomal recurrent CNVs generated by NAHR, six recurrent MEIG1 deletions were expected. However, the observed deletion frequency is significantly low (6/413 expected, 0/413 observed, P=0.015, Fisher's exact test).
There is only one CNV record (variation 23 238) from the DGV database that perfectly matched recurrent MEIG1 CNVs.26 However, there were also some other CNVs in DGV, including variations 29584,28 48486, 48487,29 53843,3 and 112611,30 the majority parts of which were overlapped with recurrent MEIG1 CNVs. Considering the fact that most of the genome-wide CNV investigating methods used in previous studies may not have fine resolution to map CNV breakpoints, we hypothesized that the above five CNVs were candidates for recurrent MEIG1 CNVs. In these published CNV data, totally 14 duplications and three deletions were reported in 4397 individuals (P=0.006, Fisher's exact test), still supporting the over representation of duplication in the MEIG1 locus.
The consistent observations in this study and previous CNV screening in human populations revealed much fewer recurrent MEIG1 deletions than expected and suggested that the MEIG1 deletion alleles were likely to be deleterious and act under purifying selection. As the murine orthologue of MEIG1 has been shown to be involved in the spermatogenesis,11, 12 a similar role of MEIG1 is promising. Therefore, the recurrent MEIG1 deletion can be a loss-of-function mutation and may affect spermatogenesis in homozygous or compound heterozygous states. It was previously reported that heterozygous Meig1-knockout mice are fertile,11 which implied that the loss-of-function mutations of MEIG1 were likely to be recessive pathogenic alleles. Although we did not identify any MEIG1 deletion in our patients with spermatogenic impairment or in healthy controls, further comprehensive studies in large cohorts and in various human populations are welcome. In addition to the MEIG1 gene, the 5' end of DCLRE1C, a gene implicated in human immune deficiency, was also affected by recurrent MEIG1 deletion.16, 17 Therefore, the purifying selection pressure can also act via human immune defects.
In conclusion, previous CNV studies in human populations identified lots of CNVs throughout the human genome, a portion of which were found to be associated with genomic disorders and other human diseases. Comprehensive CNV investigations in specific loci of interest demanded accurate and fast CNV analyzing methods, while the AccuCopy technology provided reliable and efficient assays for multiple CNV genotyping. Utilizing AccuCopy, we demonstrated that local genome architecture of SDs was an important factor underlying CNV hotspots in the human genome. Furthermore, over representation of recurrent MEIG1 duplications supported a purifying selection, via spermatogenic impairment and/or immune deficiency. The potential roles of recurrent MEIG1 CNVs in human disease were highlighted in this study and need further comprehensive investigation in the future.
References
Zhang, F., Gu, W., Hurles, M. E., Lupski, J. R. Copy number variation in human health, disease, and evolution. Annu. Rev. Genomics Hum. Genet. 10, 451–481 (2009).
Stankiewicz, P., Lupski, J. R. Genome architecture, rearrangements and genomic disorders. Trends Genet. 18, 74–82 (2002).
Itsara, A., Cooper, GM., Baker, C., Girirajan, S., Li, J., Absher, D. et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 84, 148–161 (2009).
Fu, W., Zhang, F., Wang, Y., Gu, X., Jin, L. Identification of copy number variation hotspots in human populations. Am. J. Hum. Genet. 87, 494–504 (2010).
Liu, P., Lacaria, M., Zhang, F., Withers, M, Hastings, P. J., Lupski, J. R. et al. Frequency of nonallelic homologous recombination is correlated with length of homology: evidence that ectopic synapsis precedes ectopic crossing-over. Am. J. Hum. Genet. 89, 580–588 (2011).
Bailey, J. A., Gu, Z., Clark, R. A., Reinert, K., Samonte, R. V., Schwartz, S. et al. Recent segmental duplications in the human genome. Science 297, 1003–1007 (2002).
Carvalho, C. M., Zhang, F., Lupski, J. R. Structural variation of the human genome: mechanisms, assays, and role in male infertility. Syst. Biol. Reprod. Med. 57, 3–16 (2011).
Sun, C., Skaletsky, H., Rozen, S., Gromoll, J., Nieschlag, E., Oates, R. et al. Deletion of azoospermia factor a (AZFa) region of human Y chromosome caused by recombination between HERV15 proviruses. Hum. Mol. Genet. 9, 2291–2296 (2000).
Repping, S., Skaletsky, H., Lange, J., Silber, S., Van Der Veen, F., Oates, R. D. et al. Recombination between palindromes P5 and P1 on the human Y chromosome causes massive deletions and spermatogenic failure. Am. J. Hum. Genet. 71, 906–922 (2002).
Kuroda-Kawaguchi, T., Skaletsky, H., Brown, L. G., Minx, P. J., Cordum, H. S., Waterston, R. H. et al. The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat. Genet. 29, 279–286 (2001).
Zhang, Z., Shen, X., Gude, DR., Wilkinson, BM., Justice, MJ., Flickinger, CJ. et al. MEIG1 is essential for spermiogenesis in mice. Proc. Natl Acad. Sci. USA. 106, 17055–17060 (2009).
Salzberg, Y., Eldar, T., Karminsky, O. D., Itach, S. B., Pietrokovski, S., Don, J. et al. Meig1 deficiency causes a severe defect in mouse spermatogenesis. Dev. Biol. 338, 158–167 (2010).
Lu, C., Zhang, J., Li, Y., Xia, Y., Zhang, F., Wu, B. et al. The b2/b3 subdeletion shows higher risk of spermatogenic failure and higher frequency of complete AZFc deletion than the gr/gr subdeletion in a Chinese population. Hum. Mol. Genet. 18, 1122–1130 (2009).
World Health Organization WHO Laboratory Manual for the Examination of Human Semen and Semen-Cervical Mucus Interaction, Cambridge University Press: Cambridge, (1999).
Zhang, F., Khajavi, M., Connolly, A. M., Towne, C. F., Batish, S. D., Lupski, J. R. et al. The DNA replication FoSTeS/MMBIR mechanism can generate human genomic, genic, and exonic complex rearrangements. Nat. Genet. 41, 849–853 (2009).
Moshous, D., Callebaut, I., de Chasseval, R., Corneo, B., Cavazzana-Calvo, M., Le Deist, F. et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell 105, 177–186 (2001).
Pannicke, U., Hönig, M., Schulze, I., Rohr, J., Heinz, G. A., Braun, S. et al. The most frequent DCLRE1C (ARTEMIS) mutations are based on homologous recombination events. Hum. Mutat. 31, 197–207 (2009).
Zhang, F., Lu, C., Li, Z., Xie, P., Xia, Y., Zhu, X et al. Partial deletions are associated with an increased risk of complete deletion in AZFc: a new insight into the role of partial AZFc deletions in male infertility. J. Med. Genet. 44, 437–444 (2007).
Armour, J. A., Palla, R., Zeeuwen, P. L., den Heijer, M., Schalkwijk, J., Hollox, E. J. et al. Accurate, high-throughput typing of copy number variation using paralogue ratios from dispersed repeats. Nucleic Acids Res. 35, e19 (2007).
Schouten, J. P., McElgunn, C. J., Waaijer, R., Zwijnenburg, D, Diepvens, F., Pals, G. et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 30, e57 (2002).
Shen, Y., Wu, B. L. Designing a simple multiplex ligation-dependent probe amplification (MLPA) assay for rapid detection of copy number variants in the genome. J. Genet. Genomics 36, 257–265 (2009).
International Human Genome Sequencing Consortium Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Bailey, J. A., Eichler, E. E. Primate segmental duplications: crucibles of evolution, diversity and disease. Nat. Rev. Genet. 7, 552–564 (2006).
Sen, S. K., Han, K., Wang, J., Lee, J., Wang, H., Callinan, P. A. et al. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 79, 41–53 (2006).
Han, K., Lee, J., Meyer, T. J., Wang, J., Sen, S. K., Srikanta, D. et al. Alu recombination-mediated structural deletions in the chimpanzee genome. PLoS Genet. 3, 1939–1949 (2007).
Korbel, J. O., Urban, A. E., Affourtit, J. P., Godwin, B., Grubert, F., Simons, J. F et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 318, 420–426 (2007).
Turner, D. J., Miretti, M., Rajan, D., Fiegler, H, Carter, N. P., Blayney, M. L. et al. Germline rates of de novo meiotic deletions and duplications causing several genomic disorders. Nat. Genet. 40, 90–95 (2008).
Jakobsson, M., Scholz, S. W., Scheet, P., Gibbs, J. R., VanLiere, J. M., Fung, H. C. et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 451, 998–1003 (2008).
Shaikh, T. H., Gai, X., Perin, J. C., Glessner, J. T., Xie, H., Murphy, K et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 19, 1682–1690 (2009).
Park, H., Kim, J. I., Ju, Y. S., Gokcumen, O., Mills, R. E., Kim, S. et al. Discovery of common Asian copy number variants using integrated high-resolution array CGH and massively parallel DNA sequencing. Nat. Genet. 42, 400–405 (2010).
Acknowledgements
We thank all participating subjects for their kind cooperation in the study. We also thank Dr JR Lupski for his critical review. This work was supported by the National Basic Research Program of China (2011CBA00401 and 2012CB944600); the National S&T Major Special Project (2011ZX09102-010-01); the National Natural Science Foundation of China (30930079, 31000552, 31171210 and 81100461); the Shanghai Pujiang Program (10PJ1400300); and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
ZJ is a founder of Genesky Biotechnologies.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Du, R., Lu, C., Jiang, Z. et al. Efficient typing of copy number variations in a segmental duplication-mediated rearrangement hotspot using multiplex competitive amplification. J Hum Genet 57, 545–551 (2012). https://doi.org/10.1038/jhg.2012.66
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.66
Keywords
This article is cited by
-
High leukocyte mitochondrial DNA copy number contributes to poor prognosis in breast cancer patients
BMC Cancer (2023)
-
A rare familial rearrangement of chromosomes 9 and 15 associated with intellectual disability: a clinical and molecular study
Molecular Cytogenetics (2021)
-
Association analysis of copy number variations in type 2 diabetes-related susceptible genes in a Chinese population
Acta Diabetologica (2018)
-
Copy number variants of ABCF1, IL17REL, and FCGR3A are associated with the risk of gout
Protein & Cell (2017)
-
Autism-like behaviours and germline transmission in transgenic monkeys overexpressing MeCP2
Nature (2016)
