
Abstract
Filamin A is encoded by the FLNA gene on chromosome Xq28 and functions in cross-linking actin filaments into orthogonal networks in the cortical cytoplasm. FLNA p.V528M was initially detected in a female autopsy case of X-linked bilateral periventricular nodular heterotopia (BPNH), a neuronal migration disorder characterized by subependymal nodules of gray matter. During our mutation analysis of FLNA in a boy with apparent X-linked thrombocytopenia, we detected the p.V528M variant. The patient, mother and sister, who were heterozygous for the substitution, did not have BPNH. We observed an allele frequency of 4.8% in healthy control Japanese, but did not observe the variant in Caucasian subjects. Hemizygous controls had a normal platelet count and size. We suggest that p.V528M is neither associated with BPNH nor with thrombocytopenia and giant platelets, and represents a functional polymorphism.
Similar content being viewed by others

Introduction
X-linked dominant bilateral periventricular nodular heterotopia (BPNH; MIM #300049) is a neuronal migration disorder characterized by subependymal nodules of gray matter.1, 2 Most patients are heterozygous females who suffer from epilepsy and have normal to borderline intelligence.3 Hemizygous males are likely to be embryonic-lethal or severely affected with mental retardation and skeletal dysplasia, in addition to epilepsy. X-linked BPNH is caused by mutations in FLNA (MIM #300017) on chromosome Xq28, which encodes the actin-binding protein filamin A.3 This cytoskeletal protein cross-links actin filaments into orthogonal networks in the cortical cytoplasm and participates in the anchoring of membrane proteins for actin cytoskeleton.4, 5 So far, >50 FLNA mutations have been reported as a cause of BPNH. Most are nonsense, frameshift or splicing mutations, predicted to result in a severe loss of function, whereas some missense mutations also have been identified.6, 7, 8
Filamin A is a widely expressed protein5, 9. In blood platelets, it interacts with the glycoprotein Ib/IX transmembrane receptor and contributes to the normal platelet morphology by linking the platelet membrane skeleton and cytoskeletal actin filaments.10, 11 Inherited glycoprotein Ib/IX deficiency leads to Bernard–Soulier syndrome, a rare autosomal recessive bleeding disorder exhibiting abnormally large platelets and thrombocytopenia (macrothrombocytopenia).12, 13 Defects in FLNA could possibly lead to congenital macrothrombocytopenia. During our mutation analysis of FLNA in a patient with apparent X-linked thrombocytopenia, we detected the p.V528M variant that has been reported as a BPNH mutation.14 We present evidence that p.V528M is neither associated with BPNH nor with thrombocytopenia and giant platelets.
Materials and methods
Patient
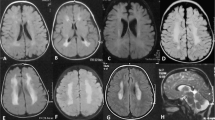
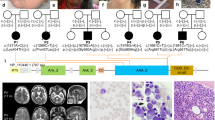
A 6-year-old boy was referred to Kobe University hospital for the evaluation of thrombocytopenia. A laboratory test revealed a platelet count of 30 × 109 l−1. Although the patient exhibited no bleeding tendency, he showed persistent thrombocytopenia (30–60 × 109 l−1; Table 1). Peripheral blood smears showed the prominent appearance of giant platelets. Other blood cell counts, blood chemistry and coagulation tests were within the normal ranges. Platelets were normally aggregated in response to ADP, collagen and ristocetin. Flow cytometry showed the normal expression of platelet glycoprotein Ib/IX.12 There were no granulocyte inclusion bodies on May–Grünwald–Giemsa (MGG)-stained peripheral blood smears, which was confirmed by immunofluorescence analysis of granulocyte myosin IIA localization.15 These results excluded Bernard–Soulier syndrome and MYH9 disorders, the most frequent forms of macrothrombocytopenia.16 The boy's maternal grandfather (I-3) had a previous history of immune thrombocytopenia and received platelet transfusion (Figure 1). Two sons (II-1and 2) of the maternal grandfather’s (I-3) sister (I-2) had thrombocytopenia, one of whom (II-2) underwent splenectomy. Based on the family history, an X-linked dominant thrombocytopenia was suggested. The parents, his sibling and maternal grandfather's sister were negative for a history of a bleeding tendency. All these individuals were of normal intelligence and did not show signs of psychotic behavior, cerebral palsy, epilepsy or other neurological disorders. Brain magnetic resonance imaging scans of the patient, mother and sister revealed a normal architecture of the brain (Figure 2).

Family tree of the patient with apparent X-linked dominant thrombocytopenia. (a) Circles represent females and squares represent males. Solid symbols denote affected and open symbols denote unaffected individuals. The proband (III-1) is indicated by an arrow. Samples were available and analyzed from II-3, II-4, III-1 and III-2. (b) Allele-specific restriction fragment length polymorphism analysis. The c.1582G>A substitution abolishes a recognition site for BstUI: the hemizygous patient displays a 547-bp band (uncut) and the wild-type father displays 215- and 332-bp bands. The heterozygous mother and sister display three bands. MW, HaeIII digest of ΦX 174 DNA; F, father (II-3); M, mother (II-4); P, patient (III-1); S, sister (III-2).

Magnetic resonance imaging of the brain. T2-weighted axial images demonstrate a normal architecture of the brain. (a) Patient (III-1); (b) mother (II-4); (c) sister (III-2).
Mutation analysis of FLNA
The entire coding regions of the patient's FLNA gene were amplified from genomic DNA by PCR and amplified DNA fragments were subjected to direct cycle sequence analysis (Supplementary Table 1). The presence of the c.1582G>A substitution was analyzed in the healthy control DNA of 192 Japanese (114 males, 78 females) and 83 Austrian Caucasians (32 males, 51 females, kind gifts from Dr Christoph Gassner, Blutspende Zuerich, Switzerland17). The gender of each control was determined by PCR detection of the SRY gene (UniSTS: 482099). DNA fragments amplified using primers FLNA105, 5′-ACCCACCAATCCTGACAGC-3′, and FLNA113, 5′-AGGCAGGAAGAGCCCATGTG-3′, were digested with BstUI (New England Biolabs, Beverly, MA, USA), electrophoresed on 2% agarose gels and stained with ethidium bromide. Expected frequencies of genotypes in the population were calculated using the Hardy–Weinberg equation.18 The 95% confidence interval of the proportion was calculated. Peripheral blood samples were obtained after obtaining a written consent. The Ethics Committees of Nagoya Medical Center and Kobe University approved this study.
Platelet size determination
Platelet size was determined as the platelet diameter by the microscopic observation of 200 platelets on MGG-stained peripheral blood smears.
Results and discussion
We performed mutation analysis of FLNA in a patient with apparent X-linked thrombocytopenia (Figure 1). The entire coding sequence of the gene including the intron–exon junctions was investigated. This revealed a hemizygous substitution, c.1582G>A, changing the conserved valine at codon 528 to methionine (p.V528M). The patient's mother and sister were heterozygous for the substitution. The c.1582G>A has been reported in a heterozygous condition in a female autopsy case of BPNH.14 In that report, the substitution was not found in 100 unrelated control individuals. Although the substitution was not found in the single-nucleotide polymorphism database (http://www.ncbi.nlm.nih.gov/SNP/, accessed May 2010), our restriction fragment length polymorphism analysis of control Japanese subjects identified four hemizygotes in 114 males and nine heterozygotes in 78 females. The allele frequency was 4.8% (95% confidence interval (2.8–8.1)). A comparison of the allele frequency between males (3.5%, 95% confidence interval (1.4–8.7)) and females (5.8%, 95% confidence interval (3.1–10.6)) showed no significant difference (P>0.05). The exact test for female data showed no significant deviation from the Hardy–Weinberg equilibrium (P>0.05). In contrast, c.1582G>A was not detected in a total of 83 control Caucasian subjects, suggesting that c.1582G>A is rare or absent in this population.
The previously described BPNH case with p.V528M reportedly had characteristic heterotopic gray matter lining the walls of the lateral ventricles on a brain magnetic resonance imaging study.14 However, the present hemizygous patient and heterozygous mother and sister had normal brain magnetic resonance imaging findings (Figure 2). FLNA is the major gene currently known to be associated with BPNH.6, 7, 8 Most FLNA mutations are nonsense, frameshift or splicing mutations, predicted to result in a severe loss of function, whereas some missense mutations have been identified. Missense mutations may result in the production of partially functional proteins and lead to milder phenotypes. A rare autosomal recessive BPNH associated with microcephaly, with mutations of ARFGEF2,19 and BPNH patients with duplication of 5p15.1 or trisomy of 5p15.3320 have also been reported. We suggest that p.V528M is not associated with BPNH and that it represents a functional polymorphism.
Although the present patient hemizygous for p.V528M had macrothrombocytopenia, hemizygous healthy control individuals showed a normal platelet count and size (21.1±2.1 × 109 l−1 and 2.7±0.4 μm, respectively, n=4). The patient's mother and sister, both of whom were heterozygous for the substitution, also had a normal platelet count and size (Table 1). We thus suggest that p.V528M does not affect the platelet count and size.
Which gene was responsible for macrothrombocytopenia in the patient? As clinical information was obtained by interview, it is not known whether thrombocytopenia in other affected males was associated with giant platelets. The most common forms of macrothrombocytopenia, that is, Bernard–Soulier syndrome and MYH9 disorders, were first excluded by flow cytometry for platelet glycoprotein Ib/IX and immunofluorescence analysis of granulocyte myosin IIA localization, respectively.16 Mutation screening of the known candidate genes for X-linked thrombocytopenia such as WAS21, 22 and GATA123 excluded the presence of mutations in the patient (data not shown). We hypothesized that the macrothrombocytopenia in the present patient was caused by the mutation of an unknown gene on chromosome X. It is also possible that thrombocytopenia and giant platelets occurred incidentally and mimicked macrothrombocytopenia.
References
Eksioglu, Y. Z., Scheffer, I. E., Cardenas, P., Knoll, J., DiMario, F., Ramsby, G. et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron 16, 77–87 (1996).
Kamuro, K. & Tenokuchi, Y. Familial periventricular nodular heterotopia. Brain Dev. 15, 237–241 (1993).
Fox, J. W., Lamperti, E. D., Eksioglu, Y. Z., Hong, S. E., Feng, Y., Graham, D. A. et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron 21, 1315–1325 (1998).
Gorlin, J. B., Yamin, R., Egan, S., Stewart, M., Stossel, T. P., Kwiatkowski, D. J. et al. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell. Biol. 111, 1089–1105 (1990).
Robertson, S. P. Filamin A: phenotypic diversity. Curr. Opin. Genet. Dev. 15, 301–307 (2005).
Parrini, E., Ramazzotti, A., Dobyns, W. B., Mei, D., Moro, F., Veggiotti, P. et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain 129, 1892–1906 (2006).
Sheen, V. L., Dixon, P. H., Fox, J. W., Hong, S. E., Kinton, L., Sisodiya, S. M. et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 10, 1775–1783 (2001).
Sole, G., Coupry, I., Rooryck, C., Guerineau, E., Martins, F., Deves, S. et al. Bilateral periventricular nodular heterotopia in France: frequency of mutations in FLNA, phenotypic heterogeneity and spectrum of mutations. J. Neurol. Neurosurg. Psychiatry 80, 1394–1398 (2009).
Feng, Y. & Walsh, C. A. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat. Cell Biol. 6, 1034–1038 (2004).
Cunningham, J. G., Meyer, S C. & Fox, J. E. The cytoplasmic domain of the α-subunit of glycoprotein (GP) Ib mediates attachment of the entire GP Ib-IX complex to the cytoskeleton and regulates von Willebrand factor-induced changes in cell morphology. J. Biol. Chem. 271, 11581–11587 (1996).
Hartwig, J. H. & DeSisto, M. The cytoskeleton of the resting human blood platelet: structure of the membrane skeleton and its attachment to actin filaments. J. Cell. Biol. 112, 407–425 (1991).
Kunishima, S., Kamiya, T. & Saito, H. Genetic abnormalities of Bernard-Soulier syndrome. Int. J. Hematol. 76, 319–327 (2002).
Lanza, F. Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy). Orphanet. J. Rare Dis. 1, 46 (2006).
Kakita, A., Hayashi, S., Moro, F., Guerrini, R., Ozawa, T., Ono, K. et al. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. Acta. Neuropathol. 104, 649–657 (2002).
Kunishima, S., Matsushita, T., Kojima, T., Sako, M., Kimura, F., Jo, E. K. et al. Immunofluorescence analysis of neutrophil nonmuscle myosin heavy chain-A in MYH9 disorders: association of subcellular localization with MYH9 mutations. Lab. Invest. 83, 115–122 (2003).
Kunishima, S. & Saito, H. Congenital macrothrombocytopenias. Blood Rev. 20, 111–121 (2006).
Kunishima, S., Gassner, C., Inoue, C., Kamiya, T. & Ozawa, K. Expression of low-frequency Ala108Pro substitution in the platelet glycoprotein Ibβ gene. Eur. J. Immunogenet. 30, 159–161 (2003).
Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Sheen, V. L., Ganesh, V. S., Topcu, M., Sebire, G., Bodell, A., Hill, R. S. et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat. Genet. 36, 69–76 (2004).
Sheen, V. L., Wheless, J. W., Bodell, A., Braverman, E., Cotter, P. D., Rauen, K. A. et al. Periventricular heterotopia associated with chromosome 5p anomalies. Neurology 60, 1033–1036 (2003).
Derry, J. M., Kerns, J. A., Weinberg, K. I., Ochs, H. D., Volpini, V., Estivill, X. et al. WASP gene mutations in Wiskott-Aldrich syndrome and X-linked thrombocytopenia. Hum. Mol. Genet. 4, 1127–1135 (1995).
Villa, A., Notarangelo, L., Macchi, P., Mantuano, E., Cavagni, G., Brugnoni, D. et al. X-linked thrombocytopenia and Wiskott-Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nat. Genet. 9, 414–417 (1995).
Nichols, K. E., Crispino, J. D., Poncz, M., White, J. G., Orkin, S. H., Maris, J. M. et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat. Genet. 24, 266–270 (2000).
Acknowledgements
This work was supported by grants from the Japan Society for the Promotion of Science (18591094 and 20591161), the Ministry of Health, Labour and Welfare (Grant for Child Health and Development 19C-2), the Mitsubishi Pharma Research Foundation and the National Hospital Organization (Network Research Grant for Congenital Thrombocytopenia).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Kunishima, S., Ito-Yamamura, Y., Hayakawa, A. et al. FLNA p.V528M substitution is neither associated with bilateral periventricular nodular heterotopia nor with macrothrombocytopenia. J Hum Genet 55, 844–846 (2010). https://doi.org/10.1038/jhg.2010.114
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2010.114
