
Abstract
Faecal microbiota transplantation (FMT) may contribute towards disease remission in ulcerative colitis (UC), but it is unknown which factors determine long-term effect of treatment. Here, we aimed to identify bacterial signatures associated with sustained remission. To this end, samples from healthy donors and UC patients—grouped into responders and non-responders at a primary end point (week 12) and further stratified by sustained clinical remission and relapse assessed at ⩾1-year follow-up were analysed, comparing the efficacy of FMT from either a healthy donor or autologous faeces. Microbiota composition was determined with a 16S rRNA gene-based phylogenetic microarray on faecal and mucosal samples, and functional profiles were predicted using PICRUSt with quantitative PCR verification of the butyrate production capacity; short-chain fatty acids were measured in faecal samples. At baseline, UC patients showed reduced amounts of bacterial groups from the Clostridium cluster XIVa, and significantly higher levels of Bacteroidetes as compared with donors. These differences were reduced after FMT mostly in responders. Sustained remission was associated with known butyrate producers and overall increased butyrate production capacity, while relapse was associated with Proteobacteria and Bacteroidetes. Ruminococcus gnavus was found at high levels in donors of failed FMT. A microbial ecosystem rich in Bacteroidetes and Proteobacteria and low in Clostridium clusters IV and XIVa observed in UC patients after FMT was predictive of poor sustained response, unless modified with a donor microbiota rich in specific members from the Clostridium clusters IV and XIVa. Additionally, sustained response was associated with restoration of the butyrate production capacity.
Similar content being viewed by others
Introduction
The complex ecosystem of the commensal microbiota and its interactions with the host are fundamental in determining health. Aberrations of the microbiota have been associated with several diseases, including inflammatory bowel disease (IBD) (Nagalingam and Lynch, 2012; Kostic et al., 2014). In ulcerative colitis (UC), the prevailing hypothesis on the pathogenesis involves either an altered composition of the microbiota that induces a pathological mucosal immune response or an excessive immune response to the normal microbiota colonizing the gastrointestinal tract (Sartor, 2006; Kaser et al., 2010). The most radical way of modifying the gut microbial ecosystem is by faecal microbiota transplantation (FMT). FMT has been shown to have a role on colitis activity in several case series (Angelberger et al., 2012; Borody et al., 2012; Kump et al., 2013; Kunde et al., 2013; Aroniadis et al., 2016), as well as three randomized controlled trials (Moayyedi et al., 2015; Rossen et al., 2015; Paramsothy et al., 2017). How and why a reset of the gastrointestinal microbiota can have beneficial effects on disease activity in only a subset of UC patients is still elusive. In addition, flares of the disease can influence the microbial environment, and this mechanism is poorly understood. Some authors state that the microbiota composition is subject to inflammation, but others have found a distinct composition in UC patients even in clinical remission (Rajilic-Stojanovic et al., 2013; Winter et al., 2013).
The microbiota of patients with IBD has been characterized by a lower abundance of Firmicutes, more specifically clostridia, Ruminococcaceae and Lactobacillus, as well as Actinobacteria such as Bifidobacterium (Ott et al., 2004; Frank et al., 2007; Kostic et al., 2014). While findings related to microbiota composition in Crohn's disease seem to be more consistent, for patients suffering from UC conflicting data have been reported. Moreover, in UC the aberrant gastrointestinal microbiota has been linked to a decrease in short-chain fatty acids (SCFAs) production, such as butyrate and propionate (Marchesi et al., 2007; Huda-Faujan et al., 2010; Tan et al., 2014). These SCFAs, and more specifically butyrate, which is mainly produced by species within the Firmicutes, are known to downregulate proinflammatory responses in intestinal epithelial cells and to modulate activity of relevant sentinel cell types (Iraporda et al., 2015). Butyrate-producing species such as Roseburia hominis and Faecalibacterium prausnitzii were found in lower abundance in faeces from UC patients as compared with healthy controls (Machiels et al., 2014). There is a wide interindividual variation in the relative abundance and composition within the Bacteroidetes phylum, and outcomes from studies comparing the abundance in faecal samples from UC patients and healthy individuals are conflicting. Nevertheless, strains of Bacteroides vulgatus and enterogenic Bacteroides fragilis (causing peritonitis or appendicitis) have been implicated in IBD, but the mechanism of action is not yet fully understood (Basset et al., 2004; Wexler, 2007). Furthermore, differences between IBD patients and healthy subjects were found not only in the gut microbial ecosystem but also in their metabolic profiles in the luminal content, which illustrates the complexity of the aetiology of IBD (De Preter et al., 2015).
Recently, we conducted a randomized controlled trial in mild to moderately active UC patients comparing the efficacy of FMT either from a healthy donor or with autologous faeces, which provides a unique human model to study the impact of changes in the microbiota on disease activity and its signatures (Rossen et al., 2015). A number of patients experienced not only a complete remission of their disease activity at 3 months but also clinical remission was sustained for at least 1 year. For this reason, the focus of this study is to identify signatures and predictors for response in both groups and their changes within the subgroup of patients who experienced long-term sustained remission, in comparison with patients who did not respond initially or relapsed within 1 year. Moreover, we studied the healthy donor microbiota to identify specific bacterial groups that are predictive for a sustained response to therapy. Finally, PICRUSt was used to predict whether microbiota functions typically associated with reduction of inflammation processes, such as butyrate production- and serpin-mediated protease inhibition capacities, were restored after FMT.
Materials and methods
Study design, subjects and sampling
Patients with mild to moderately active UC and healthy donors participated in a randomized placebo-controlled study, the TURN trial, comparing the efficacy of two duodenal infusions at a 3-week interval of faecal homogenates after bowel preparation from a healthy donor (FMT-D) or autologous faeces (FMT-A) as placebo (Rossen et al., 2015). Patients who achieved the primary end point of the trial (clinical remission; defined by a Simple Clinical Colitis Activity Index (SCCAI) ⩽2 together with a decrement of ⩾1 point in Mayo Endoscopic Score from baseline on repeat sigmoidoscopy at week 12) were called 'responders' and patients who did not achieve the primary end point were called 'non-responders'. Patients were followed for up to 3 years after inclusion and were requested to fill out an SCCAI and a questionnaire on medication use every 2 months. Sustained remission at 1 year (or more) after inclusion was defined as clinical remission (SCCAI<5) without the need for rescue therapy (Jowett et al., 2003). Relapse was defined as a SCCAI ⩾5 and/or need for rescue therapy during follow-up (FU). Patients who did not achieve the primary end point maintained the 'non-responders' classification during FU. Healthy subjects who participated in the trial as donors underwent extensive screening on bacterial, viral and parasitic stool pathogens and were classified as 'donors'.
Faecal samples were collected as described previously at baseline (t0) before bowel lavage (as preparation for endoscopy), week 12 (t12w) and 1 year or more (t⩾1 year) FU after FMT from all patients included in the trial (n=34) and were subsequently used for microbiota and SCFA profiling (Rossen et al., 2015). All patients received two FMTs (17 patients were allocated to the FMT-D group and 17 to the FMT-A group). Faeces from 15 donors were used for processing and infusion to recipients in the FMT-D group; treatments with donor faeces were performed using the same donor or different ones. For the latter, both donor samples were analysed.
Donor samples were collected at baseline (at screening of the donor) or on the day of faecal donation. If a donor donated two times for the same patient, then only one faecal sample was collected for assessment of the microbiota composition as previous studies have shown high microbiota compositional similarity over time in healthy subjects (Rajilić-Stojanović et al., 2013). In total, 24 donor samples were analysed, and those who donated for patients responding were used as healthy controls (n=10). To investigate the impact of FMT on the mucosal adherent microbiota, biopsies from the rectum were collected for microbiota profiling from UC patients during the baseline (n=29) and week 12 (n=23) by flexible sigmoidoscopy after preparation with a sodium phosphate enema. Biopsies were placed in sterile tubes immediately on the endoscopy room, snap frozen in liquid nitrogen and stored at −80 °C. There was a maximum difference of 48 h between faecal sample collection and biopsy sampling.
Microbiota profiling and functional prediction
DNA was isolated from faecal and mucosal samples as described previously (Zoetendal et al., 2006; Salonen et al., 2010). Isolated DNA was used for profiling of the microbiota using the Human Intestinal Tract Chip (HITChip), a phylogenetic microarray based on 16S rRNA gene sequences of over 1140 intestinal bacterial phylotypes (Rajilic-Stojanovic et al., 2009). The HITChip platform provides a closed system to investigate similarities and differences in microbiota composition even at low detection levels and has been benchmarked with next-generation technology sequencing (Claesson et al., 2009; van den Bogert et al., 2011). Similarity of microbiota profiles was assessed by Pearson's correlation of the probe signal intensities. Functional capacity of the microbiota was predicted based on the microbiota composition using PICRUSt (Langille et al., 2013). Using scripts in R (version 3.2.2), the species and their abundances from the HITChip were first mapped to Greengenes taxonomy from 13 May 2013 by assigning the species into OTUs with ⩾95% sequence similarity. This was then converted into BIOM format and inputted into PICRUSt v.1.0.0 (McDonald et al., 2012).
The total bacterial counts in samples was assessed by quantitative PCR (qPCR) using 0.5 ng of faecal DNA and universal bacterial primers targeting the 16S rRNA gene (Nadkarni et al., 2002). Primer concentration in each reaction was 0.2 μM. The thermal cycling conditions included an initial DNA denaturation step at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 20 s, annealing at 50 °C for 20 s, extension at 72 °C for 30 s and an additional incubation step at 82 °C for 30 s. The butyrate production capacity was determined by qPCR targeting the butyryl-coenzyme A (CoA)-CoA transferase (ButCoA) gene using 25 ng of template DNA and 0.5 μM primer concentration in each reaction as described previously (Louis and Flint, 2007). qPCR amplifications were carried out in triplicate using MX3005P Real-Time PCR System (Stratagene, La Jolla, CA, USA) in a volume of 25 μl. Each reaction was amplified by using 5 μl of HOT FIREPol EvaGreen qPCR Mix Plus, no ROX (Solis BioDyne, Tartu, Estonia).
Measurement for SCFA concentrations in faecal samples
To determine SCFAs, faecal samples were diluted in deionized water to a 10% (w v−1) concentration, followed by high-performance liquid chromatography analysis as described previously (Stams et al., 1993). Lactate, formate, acetate, propionate, isobutrate and butyrate concentrations were determined. The high-performance liquid chromatography system was equipped with a Hi-Plex H column (Agilent Technologies, Palo Alto, CA, USA) and temperature was set to 45°C at a flow of 0.9 ml min−1 running with 0.005 mM sulphuric acid as eluent and 30 mM crotonic acid as an internal standard. SCFAs were detected by determining the refractive index.
Data analysis and statistics
Demographic data were collected for all subjects included in the study and expressed as the mean and 95% confidence interval of the mean, or as the median and interquartile range (IQR) when not normally distributed. Differences between study groups (in both faecal and biopsy samples) were calculated by T-test (for parametric data) and Mann–Whitney test (for nonparametric unpaired data). Three or more group-wise comparisons were performed by Kruskal–Wallis testing at different time points on the HITChip microarray data at the genus-like level (that is, clusters of OTUs defined by a sequence similarity of 90%) between the three study groups (donors, non-responders and responders). To confirm these results, further pairwise comparisons were carried out between all groups at all time points to assess relevant signatures. A P-value <0.05 was considered statistically significant, and P-values were corrected for false discovery rate (FDR) to accommodate for multiple testing where necessary (Benjamini and Hochberg, 1995).
Statistical analyses were performed using the SPSS v. 22.0 software (SPSS, Chicago, IL, USA), R version 3.2.0 (R-package microbiome http://microbiome.github.io) and Canoco5 (ter Braak and Šmilauer, 2012). Redundancy analysis as implemented in Canoco5 was used to determine associations between the microbiota composition (130 genus-like groups included in the HITChip) and host variables (or explanatory variables) including age, gender, disease duration, site of disease, use of medication, randomization (FMT-D or FMT-A), time, subject (donor or patient), response and sustained remission (or relapse) at ⩾1-year FU. Samples at baseline, 12 weeks post FMT and 1-year post FMT were included for comparison. Significance of the explanatory variables was assessed by Monte Carlo permutation testing. Associations of signature bacteria were carried out based on redundancy analysis analyses. Further evaluations of bacterial associations mentioned in the text were carried out by corresponding appropriate analyses mentioned previously (that is, Mann–Whitney).
For the principal coordinate analysis, the genus-level data were transformed into relative abundance of total microbiota composition. Outliers in the data were defined as data points with log-transformed values >3 s.d. away from the overall log-transformed mean for each taxon. To avoid a disproportional effect of the outliers, these values were replaced by the cutoff value. Principal coordinate analysis was conducted using Bray–Curtis dissimilarities (R-package vegan). The association between the baseline principal coordinate analysis score on component 1 and response to the treatment was assessed using linear models (shown in boxplot). The analysis was conducted separately for the placebo group (FMT-A) and donor group (FMT-D).
Results
Faecal samples collected at baseline (t0, n=34) and week 12 (t12w, n=33) from UC patients were used for microbiota profiling and ButCoA gene quantification. Additionally, 27 faecal samples were collected at FU, up to 3 years after inclusion (t⩾1 years). The median FU time per group was of 17 months (IQR 12–30.7) for non-responders and 19.5 months (IQR 12–28.7) for responders with sustained remission at the t⩾1-year FU and 35 months (IQR 21.5–37) for responders that relapsed at FU. No significant differences in median FU were found for these groups (P=0.105). Study subjects characteristics are shown in Table 1.
Microbial ecosystem in UC: effects of FMT
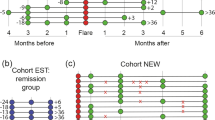
Microbiota characterization at the genus-like level revealed significant differences between healthy donors and UC patients, before and after FMT (Figure 1 and Supplementary Table 1). At baseline, UC patients showed reduced amounts of groups belonging to the Clostridium cluster XIVa as compared with donors. Interestingly, non-responders as well as those patients who responded at primary end point (t12w), but relapsed at FU, showed significantly higher levels of groups belonging to the Bacteroidetes at baseline as compared with donors, with differences ranging from 1.2- to over 11-fold higher for B. vulgatus and related species, or up to 15-fold for Prevotella species. On the contrary, patients who had sustained remission at ⩾1-year FU showed profiles more similar to those of healthy donors.

Kruskal–Wallis comparison of the 130 genus-like groups included in the HITChip between donors, non-responders and responders (the latter subdivided into patients who relapse (Rel) or sustain remission (SustRem) at ⩾1-year FU). Abundance of the different bacterial groups range from less (blue) to more (red) abundant. Red and blue outlines highlight groups either enriched or at lower levels in patients, respectively. *Genus-like groups of known butyrate-producing bacteria. Significant different groups at (a) baseline (t0, P<0.05; FDR<0.3) and (b) t12w (P<0.05; FDR<0.2).
After treatment (at t12w), differences within the Bacteroidetes remain with most patients, showing high levels as compared with donors (Figure 1). Additionally, Enterococcus species, which were found threefold higher in patient samples at baseline when compared with donors, were decreased in responders to levels similar of donors, and increased in non-responders (7.5-fold higher). Furthermore, non-responders showed significant lower levels of bacteria belonging to the Clostridium cluster XIVa, while in responders these were restored to levels similar as those found in healthy donors. Among these, we found species related to Anaerostipes caccae, Coprococcus eutactus or Eubacterium rectale along with other bacteria known to be involved in butyrate production. In addition, Proteobacteria, including potential pathogenic bacteria, were found at higher levels in non-responders as compared with donors and responders.
Similar comparisons were performed at the mucosal level at baseline and t12w between responders and non-responders, but no significant differences were observed (FDR>0.8). The microbiota composition between faecal and mucosal samples of individual patients was also compared, and differences between samples were highly reduced after therapy. This was somewhat surprising and our current explanation is that evacuation of the colon allowed the infused faecal microbiota to recolonize mucosa and lumen simultaneously (Supplementary Table 2).
Additionally, after therapy (t12w), responders in the FMT-D group showed a significantly higher similarity to their donors than non-responders (r2=0.79±0.07 and 0.70±0.09, respectively, P=0.02). This was also observed at t⩾1 year for patients with sustained remission as compared with relapsers (r2= 0.8±0.05 and 0.70±0.12, respectively), although this observation was only a trend (P= 0.1), probably due to the low number of observations in these groups. In the FMT-A group, however, no significant differences were observed between responders and non-responders similarity values with baseline samples at t12w (r2= 0.87±0.04 and 0.89±0.04, respectively) and t⩾1 year (r2= 0.82±0.04 and 0.82±0.11, respectively).
Microbial signatures of short- and long-term response to FMT
A deeper analysis of the microbiota of patients who respond to FMT showed significant differences on the overall composition of those who sustained remission as compared with those who relapsed at the ⩾1-year FU (Figure 2, FDR<0.05). Patients with sustained remission at ⩾1-year FU as well as healthy donors were found mostly positively associated to the Clostridium clusters IV (including F. prausnitzii and related species, among others) and XIVa (including Eubacterium hallii, Roseburia intestinalis or Butyrivibrio crossotus among others), all known to include butyrate-producing bacteria (Figure 2, quadrants I and II). In contrast, baseline samples of almost all patients, as well as those from donors of patients who relapsed were positively associated with the phylum Proteobacteria, including Escherichia coli or Aeromonas among other potentially pathogenic and proinflammatory bacteria (Figure 2, quadrants III and IV). Furthermore, these samples were highly associated with Ruminococcus gnavus. Albeit that in responders at t=12 weeks, the amount of R. gnavus was marginally higher than in non-responders (0.669±0.525 vs 0.548±0.856, P=0.02, FDR=0.2436), this relative abundance in responders was still low. Of note, the relative abundance at 12 weeks was 2.3-fold higher in future relapsers as compared with those who stayed in clinical remission (0.938±0.636 vs 0.400±0.170, P=0.014, FDR=0.108). Additionally, R. gnavus was found 3.8-fold higher in donors of patients who relapse as compared with donors of patients who had sustained remission (2.34±2.53% and 0.61±0.25%, respectively, P=0.08), indicative of a signature donor bacterial group prone to failure of FMT. Samples from patients who relapsed were also positively associated with groups from the Bacteroidetes phylum (including B. vulgatus and B. fragilis among others).

Redundancy analysis (RDA) of samples at t0, t12w and t1 year. Grey arrows represent the 65 best-fitting genus-like bacterial groups, shown grouped into their corresponding phyla (Bacteroides and Proteobacteria) and orders belonging to the Firmicutes phylum (Bacilli, Clostridium clusters IV and XIVa) (detailed information of bacterial groups associated with quadrants I–IV in Supplementary Table 3). Blue arrow in quadrant IV indicates R. gnavus. Twenty per cent of the variation in the data set is explained in the first two axes. (A) Samples are divided into FMT-A with sustained remission (a) and relapse (b) and FMT-D with sustained remission (c) and relapse (d) and their corresponding donors (of first or second FMT, shown as I or II). (B) Red crosses show association of explanatory variable ‘Sustained Remission’ (which includes group relapse, sustained remission and donors) with samples and bacterial groups. *Patient 8 includes two samples after 1 year. **⩾1- year sample from Patient 5 did not fulfil the quality control criteria of the HITChip pipeline.
These bacterial associations were of importance also when taking into account the source of the infused faecal sample (FMT-D and FMT-A; Figure 2). For the FMT-A treatment to be successful at the ⩾1-year FU, baseline composition needed to be closer to the ‘sustained remission’ or ‘donor’ section (e.g. patients 6 and 10). This was, however, not the case for the FMT-D treatment, where patients who showed a more disturbed baseline microbiota (that is, patients 7 and 11) could recover after treatment when transplanted with a healthy donor profile. However, patients who received FMT from a donor in the ‘relapse’ section (that is, patients 1, 2 or 5) failed to attain sustained remission. The latter observation also held for all the FMT-A patients with baseline samples in the ‘relapse’ section. Explanatory variables included in the redundancy analysis analysis such as sampling time, sustained remission and medications ‘category 1’ (that is, mesalamine, low-dose steroids or thiopurines used from week 0 until week 12) and ‘category 2’ (that is, medication 1 including high dose of steroids or anti-TNF at ⩾1-year FU) were shown to have a significant impact on the microbiota composition after FMT as calculated by Monte Carlo permutation testing (P<0.05, FDR<0.1). However, the clinical outcome and use of concomitant medication for non-responders and responders at ⩾1-year FU was not significantly different (Table 2).
Differences of response depending on the infused faecal sample were confirmed by prediction analysis (Figure 3). In the FMT-A group, we observed that patients with microbiota profiles at baseline more similar to donor samples (‘healthy’-like profiles) were more likely to respond. In this group, response was significantly associated with the known butyrate producer Coprococcus eutactus and to Ruminococcus obeum and related species (Figure 3a). Furthermore, responders and non-responders showed significantly different baseline microbiota composition (P= 0.012). However, for the FMT-D group differences in baseline microbiota between responders and non-responders with the donors was not a determining factor of response (Figure 3c).

Principal coordinate analysis (PCoA) for prediction of (sustained) response based on baseline microbiota composition for the FMT-A group (a) and the FMT-D group (c). (a) In the FMT-A group, patients with microbiota profiles at baseline more similar to donor samples (‘healthy’-like profiles, on the negative section on the x axis, highlighted in yellow) were more likely to respond (highlighted in green). This section is associated with known butyrate-producing bacteria related to Coprococcus eutactus. (b) Responders and non-responders showed significantly different baseline microbiota composition (P=0.012). Distances in baseline microbiota profiles to donor samples (score on the first principal coordinate) could also predict long-term response (FU ⩾1 year). (c) In the FMT-D group, baseline microbiota composition of patients was not a determining factor on the prediction of (sustained) response. NR, non-responders; R-Rel and R-SustRem, responders that relapse or sustain remission respectively at the ⩾1-year FU.
Butyrate in the gut: association with remission in UC
Although no significant changes were observed for single genera between responders and non-responders, summing those known to harbour butyrate-producing species showed significantly higher abundance in responders as compared with non-responders (Table 3). Hence, the microbiota profiles were used for functional prediction using PICRUSt. Among many of the functions that were significant differentially represented in the different groups (Supplementary Table 4), a significantly lower butyrate production capacity, as reflected by the butyrate-acetoacetate CoA transferase was observed in non-responders at t12w as compared with responders and donors (Supplementary Figure 1A).
To confirm the PICRUSt predictions, the effect of the treatment on the butyrate production capacity of the microbiota was determined by qPCR quantification of the ButCoA gene. Samples from donors and patients before and after treatment revealed significantly lower levels of this gene in UC patients as compared with healthy donors (Figure 4a). These levels were restored after therapy, and increased in patients who responded to the treatment, particularly in those who stayed in remission at ⩾1-year FU. In these patients, gene counts of ButCoA were increased by 6.7-fold. When comparing the relative abundance (%) of genus-like groups including known butyrate-producing bacteria in faeces, donors showed significantly higher levels as compared with non-responders at baseline and t12w; these levels were also significantly different between responders and non-responders both at baseline and t12w (Figure 4b). Similar differences were found at the mucosal level; biopsy samples of responders showed significantly higher abundance in butyrate-producing bacteria as compared with non-responders after treatment (Figure 4c). However, these results could not be confirmed by SCFA measurement in faecal samples (Supplementary Figure 2).

(a) qPCR of butyryl CoA (normalized to 16S rRNA gene copies) of donor samples and patients samples at baseline (t0) and 12 weeks after treatment (t12w). Patients are divided into non-responders (NRs) and responders, and were subdivided into those that relapse (R-Rel) or sustain remission (R-SustRem) at ⩾1-year FU. Levels of butyryl CoA were significantly lower in patients (NRs and responders) at t0 and in NRs at t12. Levels of ButCoA in responder samples with sustained remission at 1 year increase by 6.7-fold from t0 to t12w. (b and c) Relative abundance (%) of genus-like groups that include known butyrate-producing bacteria in (b) faecal samples of donors and patients at baseline (t0) and 12 weeks after treatment (t12w) and (c) in biopsy samples of patients at baseline (t0) and 12 weeks after treatment (t12w).
Since protease activity is also indicated as a mediator of inflammation, we investigated whether differences in Serpin production capacity were found between donors, responders and non-responders. Significantly, lower Serpin B levels were predicted in non-responders as compared to donors and responders, suggesting a significantly decreased protease inhibition capacity in patients not responding to the therapy (Supplementary Figure 1B).
Discussion
Our previous findings showed that FMT has an impact on the gut microbial ecosystem in UC (Rossen et al., 2015). Remarkably, these findings were observed in patients receiving healthy donor faeces but also in patients who got their own faecal microbiota, although shifts in microbiota composition after therapy were different. While patients who responded to FMT-A were more associated with an increase in Bacilli, Proteobacteria and Bacteroidetes, patients who responded to FMT-D showed an association with Clostridium clusters IV, XIVa and XVIII, as well as a reduction in Bacteroidetes.
In the current study, deeper analyses of the baseline microbiota revealed differences in patients non-responsive to FMT as compared with healthy donors, mainly within the Bacteroidetes phylum. These differences were highest for the B. fragilis and B. vulgatus genus-like groups. These bacteria have been shown to have an impact in IBD; antibodies against B. vulgatus were found in patients with Crohn's disease (Machiels et al., 2014). Moreover, B. vulgatus was found in higher abundance in faeces from ileal affected Crohn's disease patients; in UC mouse models, this bacterium was shown to invade host cells and trigger immune and proinflammatory responses (Dicksved et al., 2008; Ohkusa et al., 2009; Machiels et al., 2014). Differences between responders and non-responders were also seen in the Prevotella group (including Prevotella melaninogenica and Prevotella oralis and related species), found at higher levels in non-responders. These groups, as well as the B. fragilis group, are of special interest in the gut ecosystem, as they have been characterized as bimodal distributed bacteria and transitions between high and low abundance states of these groups can be associated with health implications as well as used for diagnostic purposes (Lahti et al., 2014). Remarkably, when further dividing responders into those that will relapse or sustain remission at ⩾1-year FU, genera from the Bacteroidetes were also found at high levels in patients who relapsed. This finding could indicate a predisposing microbiota composition to relapse in UC, high in groups belonging to this phylum at baseline, also indicative for non-response to FMT.
After FMT, and in contrast to non-responders microbiota, responders showed normalized levels of bacterial groups from the Clostridium cluster XIVa to levels similar as those found in healthy donors. Among these, we found species related to Anaerostipes caccae, Coprococcus eutactus or E. rectale along with other bacteria known to be involved in butyrate production at both the faecal and the mucosal level. In the presence of these bacteria at baseline composition was also found to be a determinant of the prediction of response, especially in patients receiving autologous FMT, while not the case for those receiving from healthy donors.
PICRUSt prediction and targeted qPCR confirmed the involvement of butyrate on response, suggesting an increase of butyrate production capacity after therapy in patients who responded to levels similar to those observed for donors. Furthermore, when these responders were stratified into those that relapsed or stayed in remission at ⩾1-year FU, we observed that the increase was mainly in those with sustained remission. Butyrate and other SCFAs are known as an energy source for colonic cells with anti-inflammatory properties (Tan et al., 2014). It has been shown to induce an anti-inflammatory response in animal models but also in clinical trials in concentrations similar to those found in the luminal space of the intestinal tract (Vernia et al., 2003; Eeckhaut et al., 2013; Iraporda et al., 2015). In an in vitro dynamic gut model, colonization with microbiota of UC patients resulted in significantly lower levels of ButCoA and butyrate-producing bacteria (Vermeiren et al., 2012). To the best of our knowledge, this is the first time butyrate production capacity is shown to have a putative role in the setting of FMT. Restoration of these bacteria as well as levels of ButCoA by FMT suggests a key role of butyrate in the long-term remission of UC, identifying butyrate or butyrate-producing bacteria as a potential treatment strategy. The finding that butyrate gene counts changes between t0 and t12 weeks was not fully paralleled by butyrate stool output may be due to various reasons, including differential gene expression, incomplete coverage of butyrate production genes, differential butyrate uptake in responders and non-responders (Boesmans et al., 2015), or trivial technical effects, all of which should be taken into account for future studies.
Similar microbial signatures of response were observed at 12 weeks post therapy. Non-responders showed significantly increased levels of Bacteroidetes and also of Proteobacteria as compared with healthy donors and responders, including potentially pathogenic bacteria such as E. coli, which was found to be twofold enriched in non-responders as compared with donors or Enterococcus spp., and over 7.5-fold higher in non-responders. Similarly to the B. vulgatus group, several studies involve E. coli with an abnormal immune and proinflammatory response in IBD (Petersen et al., 2015). Furthermore, proteases secreted from these species in faecal supernatants from UC patients have been shown to increase permeability in colonic cells, suggesting this as a possible pathogenic mechanism in IBD (Kang et al., 2010; Maharshak et al., 2015). This is supported by the significantly lower Serpin B-encoding genes predicted in non-responders microbiome.
Remarkably, none of the observed changes in the microbiota composition of faecal samples could be replicated in mucosal biopsies. Lack of significant differences in mucosal microbiota between responders and non-responders suggests that the efficacy of FMT was mainly driven by the luminal microbiota. Greater differences between faecal and mucosal samples before the therapy (baseline composition) and reduced after FMT could indicate that, after an initial effect of the therapy on the composition to a more uniform microbiota structure shared at the luminal and mucosal level, each gastrointestinal niche would recover their baseline composition, where beneficial signatures replace those associated with UC. Furthermore, it would suggest that the cross-talk between the gut–microbiota and the mucosal immune machinery is mediated via signalling molecules (for example, SCFAs), rather than a direct interaction at the mucosa adherent level. Similar observations had been reported previously, where differences in the microbiota between healthy individuals and patients suffering from irritable bowel syndrome were found to be minimal at the mucosal level as compared with differences in the faecal microbiota (Rangel et al., 2015).
In this study, a long FU period of up to 3 years after therapy allowed us to identify potential signature microbes of relapse, as well as the influence of the donor microbiota on the long-term response. As already mentioned, in responders microbiota we observed that even though considered in remission after 12 weeks, patients showing enrichment in Bacteroidetes and Proteobacteria and low levels of Clostridium clusters IV and XIVa would eventually relapse. Remarkably, relapse was also positively associated with patients who were infused with a donor microbiota with high levels (>2%) of R. gnavus (and related species), a mucolytic bacterium of the commensal microbiota (found 3.8-fold times higher than levels observed in donors from patients that experienced sustained remission). Increased levels of this bacterium have been previously reported as characteristic of the aberrant microbiota in UC (and Crohn's disease) both at the mucosal and luminal level (Png et al., 2010; Joossens et al., 2011; Berry and Reinisch, 2013). Excessive high levels of this group could be related to a higher mucolytic activity, resulting in tissue damage. We hypothesize that high amounts of R. gnavus and related species could be used as microbial predictors for unsuccessful donors. The only donor in the ‘relapse’ section associated with a positive outcome was donor-II of patient 11. However, this patient also received a previous FMT from a different donor, residing in the ‘sustained remission’ or ‘donor’ quadrant, suggesting that the imprint of the donor microbiota rather originated from the former donor. A larger study may allow assessment of whether similarity to the donor before treatment is a factor to consider when using FMT, and may help in choosing an optimal donor–patient match.
This study has several limitations: the associations of microbiota signatures and response status found are still correlative. Theoretically, it may be possible that the changes in the gut microbial ecosystem are a consequence of attenuation of disease, or the inflammation associated to it, rather than the cause of remission. However, the magnitude of signature changes of sustained responders as opposed to the changes and reversal of those who relapsed is highly suggestive of a causative mechanism. In addition, the fact that recipients with an unfavourable baseline microbiota were still capable of attaining sustained remission, only if they were transplanted with (and maintained) a donor ecosystem rich in Clostridium clusters IV and XIVa, is suggestive of a causative mechanism.
In summary, UC patients with a microbiota signature low in Clostridium clusters IV and XIVa and rich in Bacteroidetes and Proteobacteria could be predictive of poor sustained response to FMT, unless they receive a successful engraftment from a Clostridium clusters IV and XIVa-rich donor (Figure 5). Butyrate producers mainly dominate this favourable signature and durable response is associated with a restoration of the significantly reduced ButCoA gene counts found in UC patients. For donors, abundance of R. gnavus can be used as a biomarker species predictive of a lower likelihood of positive outcome. These results hold promise for novel treatment strategies, where individual intestinal microbiotas are characterized and therapies are personalized, either by targeting key bacteria or with an appropriate donor–patient match.

Proposed model of microbiota signatures in UC and healthy donors, associated with short- and long-term success of FMT.
References
Angelberger S, Lichtenberger C, Gratzer C, Papay P, Primas C, Eser A et al. (2012). P374 Fecal transplantation in patients with moderately to severely chronic active ulcerative colitis (UC). J Crohn's Colitis 6: S159–S159.
Aroniadis OC, Brandt LJ, Greenberg A, Borody T, Kelly CR, Mellow M et al. (2016). Long-term follow-up study of fecal microbiota transplantation for severe and/or complicated Clostridium difficile infection: a multicenter experience. J Clin Gastroenterol 50: 398–402.
Basset C, Holton J, Bazeos A, Vaira D, Bloom S . (2004). Are Helicobacter species and enterotoxigenic Bacteroides fragilis involved in inflammatory bowel disease? Dig Dis Sci 49: 1425–1432.
Benjamini Y, Hochberg Y . (1995). Controlling the false discovery rate—a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57: 289–300.
Berry D, Reinisch W . (2013). Intestinal microbiota: a source of novel biomarkers in inflammatory bowel diseases? Best Pract Res Clin Gastroenterol 27: 47–58.
Boesmans L, Ramakers M, Arijs I, Windey K, Vanhove W, Schuit F et al. (2015). Inflammation-induced downregulation of butyrate uptake and oxidation is not caused by a reduced gene expression. J Cell Physiol 230: 418–426.
Borody T, Wettstein A, Campbell J, Leis S, Torres M, Finlayson S et al. (2012). Fecal microbiota transplantation in ulcerative colitis: review of 24 years experience. Am J Gastroenterol 107: S665–S665.
Claesson MJ, O'Sullivan O, Wang Q, Nikkila J, Marchesi JR, Smidt H et al. (2009). Comparative analysis of pyrosequencing and a phylogenetic microarray for exploring microbial community structures in the human distal intestine. PLoS One 4: e6669.
De Preter V, Machiels K, Joossens M, Arijs I, Matthys C, Vermeire S et al. (2015). Faecal metabolite profiling identifies medium-chain fatty acids as discriminating compounds in IBD. Gut 64: 447–458.
Dicksved J, Halfvarson J, Rosenquist M, Jarnerot G, Tysk C, Apajalahti J et al. (2008). Molecular analysis of the gut microbiota of identical twins with Crohn's disease. ISME J 2: 716–727.
Eeckhaut V, Machiels K, Perrier C, Romero C, Maes S, Flahou B et al. (2013). Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut 62: 1745–1752.
Frank DN St, Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR . (2007). Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA 104: 13780–13785.
Huda-Faujan N, Abdulamir AS, Fatimah AB, Anas OM, Shuhaimi M, Yazid AM et al. (2010). The impact of the level of the intestinal short chain fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem J 4: 53–58.
Iraporda C, Errea A, Romanin DE, Cayet D, Pereyra E, Pignataro O et al. (2015). Lactate and short chain fatty acids produced by microbial fermentation downregulate proinflammatory responses in intestinal epithelial cells and myeloid cells. Immunobiology 220: 1161–1169.
Joossens M, Huys G, Cnockaert M, De Preter V, Verbeke K, Rutgeerts P et al. (2011). Dysbiosis of the faecal microbiota in patients with Crohn's disease and their unaffected relatives. Gut 60: 631–637.
Jowett SL, Seal CJ, Phillips E, Gregory W, Barton JR, Welfare MR . (2003). Defining relapse of ulcerative colitis using a symptom-based activity index. Scand J Gastroenterol 38: 164–171.
Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M et al. (2010). Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis 16: 2034–2042.
Kaser A, Zeissig S, Blumberg RS . (2010). Inflammatory bowel disease. Annu Rev Immunol 28: 573–621.
Kostic AD, Xavier RJ, Gevers D . (2014). The microbiome in inflammatory bowel disease: current status and the future ahead. Gastroenterology 146: 1489–1499.
Kump PK, Grochenig HP, Lackner S, Trajanoski S, Reicht G, Hoffmann KM et al. (2013). Alteration of intestinal dysbiosis by fecal microbiota transplantation does not induce remission in patients with chronic active ulcerative colitis. Inflamm Bowel Dis 19: 2155–2165.
Kunde S, Pham A, Bonczyk S, Crumb T, Duba M, Conrad H Jr et al. (2013). Safety, tolerability, and clinical response after fecal transplantation in children and young adults with ulcerative colitis. J Pediatr Gastroenterol Nutr 56: 597–601.
Lahti L, Salojarvi J, Salonen A, Scheffer M, de Vos WM . (2014). Tipping elements in the human intestinal ecosystem. Nat Commun 5: 4344.
Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31: 814–821.
Louis P, Flint HJ . (2007). Development of a semiquantitative degenerate real-time pcr-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl Environ Microbiol 73: 2009–2012.
Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V et al. (2014). A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63: 1275–1283.
Maharshak N, Huh EY, Paiboonrungruang C, Shanahan M, Thurlow L, Herzog J et al. (2015). Enterococcus faecalis gelatinase mediates intestinal permeability via protease-activated receptor 2. Infect Immun 83: 2762–2770.
Marchesi JR, Holmes E, Khan F, Kochhar S, Scanlan P, Shanahan F et al. (2007). Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J Proteome Res 6: 546–551.
McDonald D, Clemente JC, Kuczynski J, Rideout JR, Stombaugh J, Wendel D et al. (2012). The Biological Observation Matrix (BIOM) format or: how I learned to stop worrying and love the ome-ome. Gigascience 1: 7.
Moayyedi P, Surette MG, Kim PT, Libertucci J, Wolfe M, Onischi C et al. (2015). Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology 149: 102–109 e106.
Nadkarni MA, Martin FE, Jacques NA, Hunter N . (2002). Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148: 257–266.
Nagalingam NA, Lynch SV . (2012). Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 18: 968–984.
Ohkusa T, Yoshida T, Sato N, Watanabe S, Tajiri H, Okayasu I . (2009). Commensal bacteria can enter colonic epithelial cells and induce proinflammatory cytokine secretion: a possible pathogenic mechanism of ulcerative colitis. J Med Microbiol 58: 535–545.
Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR et al. (2004). Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 53: 685–693.
Paramsothy S, Kamm M, Kaakoush NO, Walsh AJ, van den Bogaerde J, Samuel D et al. (2017). Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet 389: 1218–1228.
Petersen AM, Halkjaer SI, Gluud LL . (2015). Intestinal colonization with phylogenetic group B2 Escherichia coli related to inflammatory bowel disease: a systematic review and meta-analysis. Scand J Gastroenterol 50: 1–9.
Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI et al. (2010). Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105: 2420–2428.
Rajilic-Stojanovic M, Heilig HG, Molenaar D, Kajander K, Surakka A, Smidt H et al. (2009). Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol 11: 1736–1751.
Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM . (2013). Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis 19: 481–488.
Rajilić-Stojanović M, Heilig HGHJ, Tims S, Zoetendal EG, de Vos WM . (2013). Long-term monitoring of the human intestinal microbiota composition. Environ Microbiol 15: 1146–1159.
Rangel I, Sundin J, Fuentes S, Repsilber D, de Vos WM, Brummer RJ . (2015). The relationship between faecal-associated and mucosal-associated microbiota in irritable bowel syndrome patients and healthy subjects. Aliment Pharmacol Ther 42: 1211–1221.
Rossen NG, Fuentes S, van der Spek MJ, Tijssen JG, Hartman JH, Duflou A et al. (2015). Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology 149: 110–118 e114.
Salonen A, Nikkila J, Jalanka-Tuovinen J, Immonen O, Rajilic-Stojanovic M, Kekkonen RA et al. (2010). Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J Microbiol Methods 81: 127–134.
Sartor RB . (2006). Mechanisms of disease: pathogenesis of Crohn's disease and ulcerative colitis. Nat Clin Pract Gastroenterol Hepatol 3: 390–407.
Stams AJ, Van Dijk JB, Dijkema C, Plugge CM . (1993). Growth of syntrophic propionate-oxidizing bacteria with fumarate in the absence of methanogenic bacteria. Appl Environ Microbiol 59: 1114–1119.
Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L . (2014). The role of short-chain fatty acids in health and disease. Adv Immunol 121: 91–119.
ter Braak CJF, Šmilauer P . (2012) Canoco Reference Manual and User's Guide: Software For Ordination, Version 5.0. Microcomputer Power: Ithaca, NY, USA.
van den Bogert B, de Vos WM, Zoetendal EG, Kleerebezem M . (2011). Microarray analysis and barcoded pyrosequencing provide consistent microbial profiles depending on the source of human intestinal samples. Appl Environ Microbiol 77: 2071–2080.
Vermeiren J, Van den Abbeele P, Laukens D, Vigsnaes LK, De Vos M, Boon N et al. (2012). Decreased colonization of fecal Clostridium coccoides/Eubacterium rectale species from ulcerative colitis patients in an in vitro dynamic gut model with mucin environment. FEMS Microbiol Ecol 79: 685–696.
Vernia P, Annese V, Bresci G, d'Albasio G, D'Inca R, Giaccari S et al. (2003). Topical butyrate improves efficacy of 5-ASA in refractory distal ulcerative colitis: results of a multicentre trial. Eur J Clin Invest 33: 244–248.
Wexler HM . (2007). Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev 20: 593–621.
Winter SE, Lopez CA, Baumler AJ . (2013). The dynamics of gut-associated microbial communities during inflammation. EMBO Rep 14: 319–327.
Zoetendal EG, Heilig HG, Klaassens ES, Booijink CC, Kleerebezem M, Smidt H et al. (2006). Isolation of DNA from bacterial samples of the human gastrointestinal tract. Nat Protocols 1: 870–873.
Acknowledgements
We thank all laboratory technicians who performed HITChip, the trial nurses involved in the TURN trial and all participants who either received or donated faeces for FMT treatments. This work was supported by MLDS Grant 2011 (WO 11-17) to NGR and NWO-Spinoza Grant 2008 to WMdV. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author contributions
CP was responsible for the study concept and design. The original study protocol was written by NR and CP. NR and CP predefined end points of the trial. CP and WdV obtained funding. NR was the executive investigator of the trial and MvdS was involved in the clinical work as a trial nurse. WdV, EZ, JH and SF were responsible for all microbiological experiments. LH and KK performed qPCR experiments on butyrate production capacity. PICRUSt experiments on functional capacity of the microbiota were carried out by JS. NR and SF performed statistical analyses. SF, NR, EZ, CP and GD drafted the manuscript. Critical revision of the manuscript was done by all coauthors. All authors had access to the study data and have reviewed and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Fuentes, S., Rossen, N., van der Spek, M. et al. Microbial shifts and signatures of long-term remission in ulcerative colitis after faecal microbiota transplantation. ISME J 11, 1877–1889 (2017). https://doi.org/10.1038/ismej.2017.44
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.44
This article is cited by
-
Human-derived bacterial strains mitigate colitis via modulating gut microbiota and repairing intestinal barrier function in mice
BMC Microbiology (2024)
-
Short-chain fatty acids: linking diet, the microbiome and immunity
Nature Reviews Immunology (2024)
-
Understanding the tonifying and the detoxifying properties of Chinese medicines from their impacts on gut microbiota and host metabolism: a case study with four medicinal herbs in experimental colitis rat model
Chinese Medicine (2022)
-
Early fecal microbiome transfer after donor defecation determines response in patients with moderate to severe ulcerative colitis
Indian Journal of Gastroenterology (2022)
-
Lactobacillus plantarum strains attenuated DSS-induced colitis in mice by modulating the gut microbiota and immune response
International Microbiology (2022)
