
Abstract
The number of microbial genomes sequenced each year is expanding rapidly, in part due to genome-resolved metagenomic studies that routinely recover hundreds of draft-quality genomes. Rapid algorithms have been developed to comprehensively compare large genome sets, but they are not accurate with draft-quality genomes. Here we present dRep, a program that reduces the computational time for pairwise genome comparisons by sequentially applying a fast, inaccurate estimation of genome distance, and a slow, accurate measure of average nucleotide identity. dRep achieves a 28 × increase in speed with perfect recall and precision when benchmarked against previously developed algorithms. We demonstrate the use of dRep for genome recovery from time-series datasets. Each metagenome was assembled separately, and dRep was used to identify groups of essentially identical genomes and select the best genome from each replicate set. This resulted in recovery of significantly more and higher-quality genomes compared to the set recovered using co-assembly.
Similar content being viewed by others

Main
Genome-resolved metagenomics involves the recovery of genomes directly from environmental shotgun DNA sequence datasets (Tyson et al., 2004). This is generally performed by assembling short-read sequences into longer scaffolds, followed by binning together scaffolds belonging to the same genomes. Metagenomic analysis of related samples from the same ecosystem is often employed to investigate compositional stability and spatial or temporal variation. The approach can also reveal microbial co-occurrence patterns and identify factors or processes that control organism abundances. Analysis of sample series data is also important technically, as different abundance patterns across the sample series for different organisms provide valuable constraints for binning (Sharon et al., 2013). In this process, reads from individual samples are mapped back to a collection of genomes that is often obtained by combining the reads from all samples and assembling them together (co-assembly; Bendall et al., 2016; Lee et al., 2017; Vineis et al., 2016). However, co-assembly dramatically increases the data set size and complexity, especially when multiple different strains of the same species are present across the sample series, and can result in fragmented assemblies (Sczyrba et al., 2017).
Independent assembly should generate more and higher-quality genomes than the co-assembly based approach because the complexity of individual samples is lower than that of the combination of samples (Supplementary Figure S1). The challenge that arises from independent assembly is that de-replication of the resulting genome set is required (Raveh-Sadka et al., 2015; Probst et al., 2016; Olm et al., 2017). De-replication involves identifying genomes that are the ‘same’ from a larger set, as well as determining the highest quality genome in each replicate set. This is important to maximize the accuracy of metabolic predictions and other downstream analyses.
De-replication requires pairwise genome comparisons, and the number of comparisons required scales quadratically with an increasing number of genomes. Hundreds of thousands of CPU hours may be needed to de-replicate larger genome sets with robust algorithms (gANI; Varghese et al., 2015). Mash, a recently developed algorithm that utilizes MinHash distance to estimate similarity between genomes, is an attractive alternative due to its incredibly fast speed (Ondov et al., 2016). However, we found that the accuracy of MASH decreases as the completeness of the compared genome bins decreases (Figure 1a). Thus, it cannot be used to de-replicate collections of partial genomes.

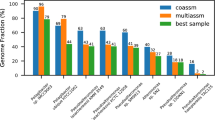
Assembly and de-replication with dRep results in more and higher-quality genome bins as compared to co-assembly. (a) A complete Escherichia coli genome was subset 10 times in increments of 10% (10%, 20%, 30% etc.). Subsets were compared to each other in a pairwise manner (100 total comparisons) using three algorithms- ANIm, MASH and gANI. For each pair of subsets, the alignment coverage between the two genomes as determined by MUMmer is shown on the x axis (aligned length / average genome length), and the ANI reported from each algorithm is shown on the y axis. ANIm and gANI are accurate when genomes are incomplete, but MASH is only accurate when genomes are essentially complete. (b) Using previously reported algorithm runtimes, we estimated the time required to de-replicate genome sets of various sizes. gANI exhibits a sharp exponential climb, limiting its use on larger genome sets; MASH and dRep do not. (c) De-replication of bins from individual assemblies and co-assembly (dRep assembly method) resulted in more bins (⩾75% complete, ⩽5% contaminated) than co-assembly alone. (d and e) Examples of genome relatedness figures generated by dRep. The red dotted line is the value of the lowest ANI resulting from a self-vs-self alignment of each genome in the cluster.
Here we present dRep, a program that utilizes both gANI and Mash in a bi-phasic approach to dramatically reduce the computational time required for genome de-replication, while ensuring high accuracy. The genome set is first divided into primary clusters using Mash, and then each primary cluster is compared in a pairwise manner using gANI, forming secondary clusters of near-identical genomes that can be de-replicated. Using published information about time required for genome comparisons, we performed an in silico simulation of de-replication time for Mash, gANI, and dRep (Figure 1b). The results indicate that dRep affords a multiple orders of magnitude increase in computation efficiency compared to naïve gANI.
To verify this prediction and to test dRep’s accuracy, we ran dRep on 1,125 genomes assembled from 195 fecal metagenomes collected from 21 premature infants during the first months of life (Raveh-Sadka et al., 2016). Genomes were 50–100% complete and contained between 0% and 24% contamination according to checkM (Parks et al., 2015). dRep clustered genomes in an identical manner to naïve gANI using default parameters, and showed near-perfect precision and recall using a variety of other parameters (Supplementary Table S1). Mash, on the other hand, resulted in a recall of 51.3% and a precision of 99.9% when compared to gANI, consistent with underestimation of similarity between incomplete genomes (Figure 1a). The actual run times were also very close to those predicted by our simulation: 92 versus 93 CPU hours for dRep, 2673 vs 2784 CPU hours for naïve gANI (actual vs predicted run times), and <1 CPU hour in both cases for Mash. As the run-time of dRep depends on the diversity of the genome set, and pre-term infant gut communities are especially non-diverse (Gibson et al., 2016; Ward et al., 2016), even greater increases in computational efficiency are expected from most other environments than predicted by our simulation.
We analyzed the same 195 metagenomes to test the prediction that, for each infant, individual assembly and de-replication would generate more and higher-quality genomes than co-assembly of the read datasets. We de-replicated genomes obtained from assemblies generated from each sample individually as well as from a co-assembly (to recover low-abundance genomes), and recovered a de-replicated genome set with 39% more bins (⩾75% complete, ⩽5% contaminated) than were obtained from co-assembly alone (270 vs 194 genomes; Figure 1c). 76 bins were recovered only from individual assemblies, 35 only from co-assemblies, and 159 from both methods.
We next compared genomes recovered using both methods. A Wilcoxon signed-ranks test indicated that scaffold length, as measured by N50, was significantly higher in genomes from dRep (median=34 046 bp) than genomes assembled from co-assemblies (median=26 103 bp), P=4.0e−11. Completeness was also significantly higher in genomes from dRep than co-assembly overall (P=6.0e−8), and although the median value was the same for genomes from both sets (median=98.3%), the 5% quantiles were different (91.6% vs 84.9%, respectively; Supplementary Table S2). Visualizations of the similarity between groups of genomes were also generated using dRep (Figures 1d and e; Supplementary Figure S2). These may be particularly valuable for comparing the population structures of groups of genomes. Taken together, dRep enabled recovery of more and better genomes than co-assembly alone, and is an effective tool for exploring the similarity among large set of genomes.
We used a published fecal metagenome data set with known strain heterogeneity to explore the effect of within-population variation on assembly and genome recovery (Sharon et al., 2013). Samples from the single infant were either co-assembled or samples were assembled individually and then de-replicated using dRep. In the case of Staphylococus hominis, co-assembly generated a contaminated bin (that is, many duplicate and triplicate single copy genes; Figure 2a). In contrast, a near-complete, uncontaminated genome was recovered from several individual time-points. Previous work on the same data set (Eren et al., 2015) has shown manual bin curation of the co-assembled bin with anvi’o can increase the S. hominis bin quality (73% complete; 6.6% redundant), but still not to the level of the un-curated bin from the individual assembly (98% complete; 0% redundant).

Strain heterogeneity reduces genome assembly quality and causes fragmentation in areas of extensive population-level variation. (a) Compared to individual assembly, co-assembly resulted in many duplicate and triplicate single copy genes. (b–d) The Staphylococcus aureus bin obtained from co-assembly is more fragmented than that from an individual assembly. (b) Scaffolds from both bins are aligned to a complete reference genome (2.7 Mbp). (c) Scaffolds from the co-assembly are aligned to a single scaffold (shown in gray in b) from the individual assembly. (d) Reads from all samples aligned to a gap in the alignment in c. Reads mapped to the area where co-assembly failed to recover a genome sequence (highlighted in blue) show signs of population-level strain variation. Gray boxes represent reads, and colored lines represent discrepancies between reads and reference sequence.
For Staphylococcus aureus, both co-assembly and individual assembly resulted in near-complete and uncontaminated genomes. However, alignment of the scaffolds from both S. aureus assemblies to a complete S. aureus reference genome showed that the genome from the co-assembly was more fragmented than that from the single sample assembly. (Figures 2b and c). Fragmentation was also concentrated in areas of extensive population variation, as evident based on SNPs between metagenome reads and the genome sequence (Figure 2d). Genome fragmentation in sites of elevated strain variation could systematically decrease measures of within-population heterogeneity that rely on mapping reads to reconstructed genome sequences (Bendall et al., 2016; Quince et al., 2016).
It is both logical, based on the well known effects of sample complexity, and clear from the analysis of human microbiome samples presented here, that assembly of data from individual samples followed by de-replication has major advantages over co-assembly (especially as co-assembled genomes can be included in the de-replication process). Because it relies on Mash, dRep can only be used if the genomes in the comparison set are >50% complete. dRep combines checkM for completeness-based genome filtering (Parks et al., 2015), Mash (Ondov et al., 2016) for fast grouping of similar genomes, gANI (Varghese et al., 2015) or ANIm (Richter and Rosselló-Móra, 2009) for accurate genomic comparisons, and Scipy (Jones et al., 2001) for hierarchical clustering. In the case of viruses and plasmids, dRep requires use of an independent method to estimate genome completeness because there are currently no established metrics for this in checkM.
dRep is easy to use, highly customizable, and parallelizable. If desired, dRep can perform rapid pairwise genomic comparisons (without de-replication) to enable visualization of the degree of similarity among groups of similar genomes (Figure 1; Supplementary Figure 2). Version 0.5.5 of dRep is available in the Supplementary Information section (Supplemental Data 1 and 2), and for up-to-date source-code, installation instructions, and the manual, see https://github.com/MrOlm/drep.
References
Bendall ML, Stevens SL, Chan L-K, Malfatti S, Schwientek P, Tremblay J et al. (2016). Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. ISME J 10: 1589–1601.
Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML et al. (2015). Anvi’o: an advanced analysis and visualization platform for ‘omics data. Peer J 3: e1319.
Gibson MK, Wang B, Ahmadi S, Burnham C-AD, Tarr PI, Warner BB et al. (2016). Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat Microbiol 1: 16024.
Jones E, Oliphant T, Peterson P . (2001). SciPy: Open source scientific tools for Python. Available at: httpscipy.org.
Lee STM, Kahn SA, Delmont TO, Shaiber A, Esen ÖC, Hubert NA et al. (2017). Tracking microbial colonization in fecal microbiota transplantation experiments via genome-resolved metagenomics. Microbiome 5: 50.
Olm MR, Brown CT, Brooks B, Firek B, Baker R, Burstein D et al. (2017). Identical bacterial populations colonize premature infant gut, skin, and oral microbiomes and exhibit different in situ growth rates. Genome Res gr-213256 27: 601–612.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S et al. (2016). Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol 17: 132.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW . (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055.
Probst AJ, Castelle CJ, Singh A, Brown CT, Anantharaman K, Sharon I et al. (2016). Genomic resolution of a cold subsurface aquifer community provides metabolic insights for novel microbes adapted to high CO2 concentrations. Environ Microbiol 19: 459–474.
Quince C, Connelly S, Raguideau S, Alneberg J, Shin SG, Collins G et al. (2016), De novo extraction of microbial strains from metagenomes reveals intra-species niche partitioning. Available at: http://biorxiv.org/lookup/doi/10.1101/073825 (Accessed 12 September 2016).
Raveh-Sadka T, Firek B, Sharon I, Baker R, Brown CT, Thomas BC et al. (2016). Evidence for persistent and shared bacterial strains against a background of largely unique gut colonization in hospitalized premature infants. ISME J 10: 2817–2830.
Raveh-Sadka T, Thomas BC, Singh A, Firek B, Brooks B, Castelle CJ et al. (2015). Gut bacteria are rarely shared by co-hospitalized premature infants, regardless of necrotizing enterocolitis development Kolter R (ed). eLife 4: e05477.
Richter M, Rosselló-Móra R . (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131.
Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Droege J et al. (2017). Critical Assessment of Metagenome Interpretation − a benchmark of computational metagenomics software. BioRxiv, 099127. Available at: https://doi.org/10.1101/09.
Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF . (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23: 111–120.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43.
Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC et al. (2015). Microbial species delineation using whole genome sequences. Nucleic Acids Res 43: 6761–6771.
Vineis JH, Ringus DL, Morrison HG, Delmont TO, Dalal S, Raffals LH et al. (2016). Patient-specific Bacteroides genome variants in pouchitis. mBio 7: e01713–e01716.
Ward DV, Scholz M, Zolfo M, Taft DH, Schibler KR, Tett A et al. (2016). Metagenomic sequencing with strain-level resolution implicates uropathogenic E. coli in necrotizing enterocolitis and mortality in preterm infants. Cell Rep 14: 2912–2924.
Acknowledgements
Funding was provided by the Sloan Foundation (http://www.sloan.org/, grant number: G 2012-10-05, PI: JFB) and the National Institutes of Health (NIH; award reference number 5R01-AI-092531). This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1106400.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Olm, M., Brown, C., Brooks, B. et al. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J 11, 2864–2868 (2017). https://doi.org/10.1038/ismej.2017.126
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.126
This article is cited by
-
Deciphering the gut microbiome of grass carp through multi-omics approach
Microbiome (2024)
-
Biofilm colonization and succession in a full-scale partial nitritation-anammox moving bed biofilm reactor
Microbiome (2024)
-
Weathered granites and soils harbour microbes with lanthanide-dependent methylotrophic enzymes
BMC Biology (2024)
-
Cataloging the phylogenetic diversity of human bladder bacterial isolates
Genome Biology (2024)
-
Particle-attached bacteria act as gatekeepers in the decomposition of complex phytoplankton polysaccharides
Microbiome (2024)
