
Abstract
The great majority of microbial species remains uncultured, severely limiting their taxonomic characterization and thus communication among scientists. Although Candidatus was devised as a provisional category to classify uncultured taxa, it has not been widely accepted owing to technical limitations and lack of priority of Candidatus names in the official nomenclature. High-throughput sequencing provides the potential for data-rich taxonomic descriptions of uncultivated microbes, comparable in quality to those of cultured organisms. In order to fully realize this potential, standards and guidelines on how to perform these descriptions are needed. Here we aimed to outline these standards and draw the roadmap for a new genome-based taxonomy that, at least initially, would be parallel but highly convergent to the one in existence for isolates. In particular, we recommend the use of DNA genome sequences, recovered by population binning or single-cell techniques, as the basis for (i) identification and phylogenetic placement, (ii) bioinformatics-based functional and thus phenotypic predictions, as well as (iii) type material. We also recommend the implementation of an independent nomenclatural system for uncultivated taxa, following the same nomenclature rules as those for cultured Bacteria and Archaea but with its own list of validly published names. If widely adopted, this system will not only facilitate a comprehensive characterization of the ‘uncultivated majority’, but also provide a unified catalogue of validly published names, thereby avoiding synonyms and confusion. We also suggest that a committee of experts, supported by an international microbiological society, should be formed to govern the new classification system.
Similar content being viewed by others

Introduction
The exact number of bacterial and archaeal species (that is, the prokaryotes) on the planet remains an unresolved issue of considerable debate with estimates ranging from millions to trillions (Amann and Rossello-Mora, 2016; Locey and Lennon, 2016; Schloss et al., 2016). Yet, there is no doubt that the great majority remains unclassified as only about 13 000 species of Bacteria and Archaea have been described and their names validly published to date (http://www.bacterio.net/; Parte, 2014). This represents a major limitation for better understanding, studying and communicating about the biodiversity on the planet. The main underlying reason is that, in recent times, the current classification has been limited to pure cultures, although the great majority of microorganisms cannot yet be cultured under laboratory settings. In fact, there is no official classification but just an official nomenclature that is ruled by the International Code of Nomenclature of Prokaryotes (ICNP; Parker et al., 2015), under the auspices of the International Committee for Systematics of Prokaryotes (ICSP). A new taxonomic name will be validly published, thus officially recognized, only if the description includes the deposition of pure cultures in two international culture collections. In the early 1990s, the development of rRNA-based molecular techniques applied to the identification and visualization of uncultured organisms promoted the establishment of a provisional taxonomic status that was called Candidatus (Murray and Schleifer, 1994; Murray and Stackebrandt, 1995). Description of a Candidatus required a 16S rRNA gene sequence assigned by a specific oligonucleotide probe to microbial cells visualized microscopically and, subsequently, further characterization by features such as morphology, Gram or other cell staining, habitat localization, unusual cellular features and a growth temperature estimate inferred from the habitat. This provisional category was adopted only by a few microbiologists often working with endosymbionts (for example, Collingro et al., 2005) or microorganisms with conspicuous features, such as magnetotactic bacteria (for example, Abreu et al., 2007) or giant sulfur bacteria (Salman et al., 2011).
Furthermore, the Candidatus status has never been covered by the ICNP, therefore Candidatus names are not validly published and have no priority. This also means that the name would not necessarily be retained in the case of isolation and formal description of another representative of the Candidatus taxon. The lack of standing in nomenclature has discouraged scientists in establishing general standards to classify uncultured taxa and resulted in a lack of a clear record of what has been hitherto classified as Candidatus. The number of Candidatus catalogued in the List of Prokaryotic Names with Standing in the Literature (http://www.bacterio.net/index.html) is around 360, but there are certainly many additional names that have never been catalogued. The lack of standing of Candidatus in the ICNP also resulted in a lack of a nomenclatural review of the new classifications and consequently errors in the etymologies of about 30% of the names (Oren, 2016). Such errors would impede the valid publication of the corresponding taxon in the case of a formal classification once a culture is available.
In addition, most of the uncultured taxa newly discovered in the past decades have not been given Linnean binominal names but just received simple alphanumeric identifiers, such as clades Ia and Ib of the surface SAR11 oceanic bacteria. These identifiers are neither descriptive of phenotypic or ecological features nor indicative of taxonomic categories, such as phylum, class, order, family, genus or species. Finally, redundant identifiers for the same taxon are common nowadays, as a regulation of the alphanumeric identifier has been lacking, causing confusion in communication among scientists of Babylonian dimensions. For instance, an abundant group of gammaproteobacterial sulfur-oxidizing bacteria is referred to in literature as either GSO or SUP05 (Lavik et al., 2009; Glaubitz et al., 2013). Marine waters are often dominated by two taxa of Alphaproteobacteria, one referred to as SAR11 and the other as the Roseobacter clade. Few ecologists might be aware that SAR11 refers to a large monophyletic group of a depth similar to a phylum, whereas the Roseobacter clade encompasses a single family (Yarza et al., 2014). Another example of the lack of generalized criteria is Prochlorococcus marinus, the most abundant photosynthetic cyanobacterium in the open ocean (Coleman and Chisholm, 2010; Luo and Konstantinidis, 2011). This ‘species’ is as diverse in sequences as many classified families. The proposal outlined here aims to address all these limitations and thus facilitate standardized taxa descriptions for uncultured organisms and communication among researchers.
High-quality taxon descriptions should be primarily based on thorough analyses of phylogenetic affiliation, genomic coherence and phenotypic characters to reveal uniqueness in the framework of the classification system, that is, no exact match in the classification (Rossello-Mora and Amann, 2015). The molecular biological tools applied to circumscribe taxa have enormously improved during the past two decades, especially after the development of the next-generation sequencing approaches, leading to relatively solid classifications based on numerical identity boundaries obtained by pairwise comparisons of genes (especially 16S rRNA and other housekeeping genes) and whole genomes (Rossello-Mora and Amann, 2015). In contrast to the rapid progress in describing genotype, the approaches to characterize phenotype continue to be cumbersome and unreliable, often to a degree that the traits determined are uninformative or irrelevant (Sutcliffe, 2015; Whitman, 2015). As a consequence, the taxonomic descriptions are more and more based on genetic and genomic circumscriptions. Conspicuously, bioinformatics-based functional predictions from the whole-genome sequence might hold higher potential for standardizing descriptions of the phenotype of the organism than the current practice.
We foresee that the dominant criteria of species and genus circumscriptions will be pairwise comparisons of genomes to define genetic discreteness and to assess low taxonomic rank classification (for example, family and below), accompanied by phylogenetic reconstructions based on 16S rRNA gene sequences to assess (mostly) high ranks (for example, order and above), and phenotype assessment based on bioinformatics functional gene annotation. For instance, the genome-aggregate average nucleotide identity (ANI), that is, the mean identity of all shared genes between two genomes, has been shown to correlate tightly with DNA–DNA hybridization, the ‘gold standard’ method for circumscribing species (Goris et al., 2007). By coupling the average amino-acid identity (AAI) and the ANI with the 16S rRNA gene phylogeny approaches, one can robustly assess the taxonomic rank and placement in the tree of life of a query genome (Konstantinidis and Tiedje, 2005). As a result of the technical improvements and the growing preponderance of the genomic information to circumscribe taxa, a proposal to ICSP has recently been made to use the DNA sequence as the type material for new classifications (Whitman, 2015). Allowing genome sequences uploaded in public repositories to serve as the type material, especially for uncultivated microbes, would greatly facilitate the digitalized, faster and less complex procedure to classify these organisms. And, most importantly, if widely adopted, this approach would advance the standing in nomenclature of the classified microorganisms that cannot yet be readily grown in pure culture in the laboratory, thus their names would be amendable to a valid publication. However, as long as the ICNP does not change or extend the nature of the required type material, it is not possible to validly publish the names of uncultivated Bacteria and Archaea.
In the past decade, improvements of sequencing techniques and bioinformatics tools have revolutionized the way uncultured microorganisms can be classified. Binning of single-population genomes from metagenomes and the technology to amplify parts of the genome from single cells are nowadays dominating the description of candidate taxa, moving it from a data-poor past to a data-rich future (Konstantinidis and Rosselló-Móra, 2015). The metabolic potential of novel candidate taxa needs no longer to be extrapolated from phylogenetic affiliation but can be more reliably deduced from genomes. The ease of sequencing will soon provide ample spatiotemporal information on the taxon’s occurrence in the environment. Therefore, we propose here a procedure that will allow the rapid, yet accurate descriptions of members of the uncultivated majority. We would also like to encourage microbial ecologists to create their own classification by defining standards and supervising a list of validly published names of uncultivated taxa. This taxonomy should be aimed to converge with that of the cultured microorganisms in a not-so-distant future.
Standards suggested to describe uncultivated taxa
Our proposal is based on the concept of species as a discrete, monophyletic and genomically as well as phenotypically homogenous population of organisms that can be discriminated from other related populations by means of diagnostic properties (Rossello-Mora and Amann, 2001; Stackebrandt et al., 2002). Monophyly is today readily determined by comparative sequence analysis of housekeeping genes such as the 16S rRNA gene and genetic discreteness is typically assessed by pairwise genomic determinations, originally DNA–DNA hybridization values, which are currently being replaced by ANI values calculated from genome sequence data. An important issue remaining is the phenotype where for uncultured microorganisms we propose to move towards standardized prediction by automated annotation of genome data and—wherever possible—the validation of these hypotheses by single cell or other methods that can assess function. We would like to argue that the latter approach, which can be equally applied to cultured and uncultured organisms, is not inferior to the non-standardized phenotyping currently in use (Sutcliffe, 2015). We are convinced that a classification of yet uncultured taxa based on binning of metagenomic data or single-cell genomics can be as stable and reliable as that established for cultured Bacteria and Archaea (Konstantinidis and Rosselló-Móra, 2015).
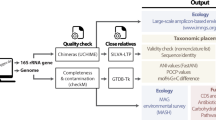
In order to make the classification of uncultured species widely applicable, we propose a set of minimum standards and a set of additional, highly recommended data types that should be obtained, whenever possible, in order to provide richer descriptions and facilitate future research. Minimum standards for descriptions of uncultured species should include the following items: (i) a complete or almost complete genome sequence (>80% completeness; <5% contamination) as a basis for a detailed analysis of genomic discreteness against their closest relatives (for example, ANI/AAI values) and a prediction of metabolic traits based on functional gene annotation; (ii) ecological data in which habitat(s) the organism is found, how abundant the organism is within the habitat and how stable its population may be over time or space (for example, to distinguish transient, allochthonous organisms from autochthonous ones). Additional data that could be obtained for richer descriptions include: (iii) the complete or almost complete 16S rRNA gene sequence for a reliable phylogenetic affiliation of the organisms in a global framework and phylogenetic probe design; (iv) experimental data confirming the bioinformatics predictions related to the key metabolic functions of the organisms; and (v) a picture of the organism derived through microscopy and fluorescence in situ hybridization. Having a picture of the organism and some experimental validation of the bioinformatics predictions related to key metabolic properties provides valuable information for future research. However, in some cases, owing to low cellular rRNA contents or impermeable cell walls, identification by fluorescence in situ hybridization might fail (for example, Luef et al., 2015), metabolic activity might be too low to measure in situ or the 16S rRNA gene sequence may not be assembled from complex metagenomes. Therefore, the standards (iii)–(v) above should be highly recommended but not mandatory (Figure 1).

Flow diagram summarizing how the data required for high-quality descriptions of uncultivated taxa can be obtained. Representative bioinformatics software or technology to use for each task is shown on the connecting lines. Dashed lines denote data or tasks that could be omitted for minimum quality descriptions (see text for details). HISH-SIMS, halogen in situ hybridization and secondary ion mass spectroscopy; MAR-FISH, microautoradiography and fluorescence in situ hybridization; MLSA, multilocus sequence analysis.
All descriptions of uncultured taxa that do not meet these standards should not be taken into account for an official valid publication of their names but should instead be deposited in the public domains, using alphanumeric identifiers, in order to facilitate their analysis and future taxa descriptions (for example, searching new genome sequences against them). We believe that the above standards are flexible enough to be achievable for most organisms and hence can represent a broadly applicable, yet robust foundation for a classification system suited for all microorganisms, not only the uncultivated ones. Below we discuss in more detail the key standards of this proposal.
Owing to the nature of the bioinformatics tools, binned genomes from metagenomes typically constitute a mosaic of the different genotypes of a single population coexisting in the same environment. This has the advantage that the bin represents a ‘consensus’ of the population and the disadvantage that a single unique genome may not be retrievable. A high-quality genome bin may be defined as being, at minimum, ⩾80% complete and containing <5% chimeric sequences and it should be linked to an almost complete sequence of the 16S rRNA gene (>1400 bp). The completeness and level of contamination can be estimated by the presence–absence and multiple copies of the universal genes, respectively, performed either manually (Albertsen et al., 2013) or with tools, such as CheckM and the HMM.essential.rb script of the enveomics collection (Parks et al., 2015; Rodriguez and Konstantinidis, 2016). The above genome-quality standards have been shown to work well with the latter methods and tools in our hands and the experience of others (Albertsen et al., 2013; Rodriguez-R and Konstantinidis, 2014). For instance, CheckM analysis of complete prokaryotic genomes available in RefSeq database identified several genome sequences with completeness values <90% due to being highly divergent and thus not providing reliable matches for some of the universal protein-coding genes (Rodriguez-R and Konstantinidis, unpublished). Assessment of the quality of the genome assembly and bioinformatically inferred functional annotations of the gene content of the genome should follow the minimum standards for genome descriptions established by the Genomic Standards Consortium (Field et al., 2008), which can be similarly applied for uncultivated taxa based on their genome sequences. The bioinformatics descriptions should include, as a minimum, the methods for recovering the genome sequence and evaluating its completeness, how functional gene predictions were performed (software, databases used and o on) and how the organisms are predicted to make a living, for example, key metabolic pathways present and degree of relatedness to experimentally determined homologous pathways. We also suggest that at least one-third of the genes in the genome are reliably annotated with functions other than hypothetical as a minimum standard of functional gene prediction. The description should also include any conspicuous or unique properties related to genome structure (for example, extremely low or high guanine–cytosine percentage) or physiological adaptation (for example, amino-acid substitutions and/or gene content selected by extremely low pH habitats). Further, there is a tight correlation between AAI and percentage of the genes in genome shared; thus the two variables are not independent from each other and AAI values are good predictors of functional gene content similarity (Konstantinidis and Tiedje, 2005). However, in cases where the genomes deviate from this correlation or there are genes of special interest for the phenotype of the organism(s) under study, the gene content differences should be discussed in detail and possibly guide the taxonomic descriptions. Any trait that may be considered strain-specific and not belonging to the core genome, such as those encoded by plasmids, must not be taken into account as diagnostic features for the new taxon. Such traits can be identified, for instance, by comparison of genomes of the population obtained by assembly of different samples or single-cell amplified genomes (SAGs) techniques or by lower coverage in the read recruitment plots compared with the rest of the genome.
For genomic discreteness, the ANI/AAI standards that have been shown to work best thus far should be used, that is, a new species typically has less than about 95% ANI with its closest relatives and a new genus has less than about 65% AAI with related genera (Goris et al., 2007). It is also important to note that ANI/AAI values can be accurately estimated even for draft (incomplete) genomes, based on as few as ~100 randomly distributed genes across the genome or ~4% of the genome (Konstantinidis et al., 2006; Richter and Rossello-Mora, 2009). Such analyses can be readily applied to draft genomes and those derived from SAGs or population bins recovered from metagenomes. Other comparable whole-genome-derived measurements such as single-nucleotide polymorphism or k-mer analysis will also be adequate. Phylogenetic affiliation, monophyly of the members of the same taxon and taxonomic rank assessment at genus level and above should be addressed by comparative sequence analysis of the 16S rRNA gene, as is the common practice nowadays, unless 16S is not available, and the genome sequence such as genome-derived AAI values (Table 1). For the past decade, a cutoff value of about 98.6% (Stackebrandt and Ebers, 2006) using the almost full-length sequence to minimize the effect of sequencing errors (Yarza et al., 2014), has been accepted as the highest identity value that generally guarantees that two organisms belong to distinct species (despite that different species could sometimes share values above this threshold). A more conservative 16S rRNA gene identity cutoff of 97% had been in use previously (Stackebrandt and Goebel, 1994), often leading to the underestimation of the species diversity (Yarza et al., 2014). Higher taxa boundaries are spaced by steps of approximately 3%, so that pairwise 16S identity values of <95%, <92%, <89%, <86% and <83% are indicative of affiliation with different genera, families, orders, classes and phyla, respectively (Rossello-Mora and Amann, 2015). The corresponding values for AAI are <45%, 45–65% and 65–95% for (different) family, genus and species, respectively (Table 1). Note that the 16S rRNA gene and AAI/ANI values proposed for defining taxonomic ranks are approximate, not absolute, thresholds and should be adjusted, as necessary, in order to better capture phenotypic or ecological distinctiveness of the candidate taxa, as was also suggested for DNA–DNA hybridization. For instance, several natural populations show ⩾97–98% intrapopulation genetic relatedness because they presumably represent younger entities. Furthermore, it is possible that distinct subpopulations (ecotypes), which have not differentiated enough genomically or ecologically to be easily discernible based on phylogenetic or read recruitment plot analysis, may exist within sequence-discrete populations, and more detailed analysis will be typically required to elucidate such subpopulations (for a more extensive discussion of these issues, see Caro-Quintero and Konstantinidis, 2012).
When a genome is derived from SAGs (Ishoey et al., 2008), which represent the same sequence-discrete population (Caro-Quintero and Konstantinidis, 2012), ideally, the consensus genome sequence of several SAGs should be provided together with the individual SAG sequences. Such a co-assembly of the SAGs would be similar to a metagenomic bin and would circumvent the limitation of individual SAGs to recover only parts of the genome, often <50% of the genome. SAGs often recover the full rRNA operon, which is commonly missed during shotgun sequencing of complex communities as assembly fails. In our view, the availability of 16S sequence is very valuable for the new system (discussed above) and comparisons against the system for isolates and thus recovery of its full-length sequence should be aggressively pursued by employing methods, such as SAGs, PCR-walking and epicPCR (Spencer et al., 2016). Descriptions based on a single SAGs should be avoided just as newly cultured species should not be described based on a single isolate.
Supervising and managing an official classification of candidate taxa
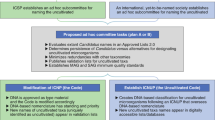
The fact that the ICNP does not oversee Candidatus taxon descriptions and thus the names cannot be validly published nor have priority, together with the lack of a generally accepted strategy to classify new candidate taxa reinforces the need to take action, otherwise the majority of uncultivated taxa will remain unclassified for the foreseeable future. We believe it is high time for microbial ecologists to establish their own official committee that will make recommendations on how to classify uncultured taxa with harmonized high standards, supervise and manage an ‘official’ classification and the rules of the nomenclature of uncultured taxa. The standards for the description, layout of the protologues (see below), data storage and, most importantly, the nature of the type material should be governed by the new committee. The standards proposed here (for example, Table 1) represent what works best in our experience with the technologies that are currently available. Obviously, these standards could change in the future with new technological advancements or the recommendations of the governing committee.
For convergence with the nomenclatural system for cultured microorganisms, the candidate species descriptions must follow the binomial nomenclature and the standards of the ICNP (with the exception of the nature of the type material), under the supervision of etymology experts. For practical reasons such as to quickly distinguish uncultivated taxa from their cultured counterparts by their names and to deviate the least from the system for isolates, we suggest to highlight the names of uncultivated taxa with a simple prefix such as a U superscript (for example, UPseudomonas atlantica, all in italics, Candidatus should not be used), which would be omitted once the organism is brought into culture (for example, Pseudomonas atlantica). We believe that each taxon description should be accompanied by an electronic voucher, as it is starting to be required in some journals publishing new taxa (Rossello-Mora et al., 2017), in the form of a digitalized protologue, describing the associated metadata such as origin and physicochemical conditions of the sample, etymology, accession links to genome and (meta)genome sequences, as well as how the genome was recovered and evaluated (for example, software and parameters used). Although some of this information is currently captured by public databases such as NCBI and EMBL, the information available in these databases is not systematic, does not typically cover ecological and taxonomic aspects adequately and, perhaps more importantly, it is not easily searchable. Standardizing and digitalizing (for example, protologue) the classification of uncultivated microorganisms will have many benefits; most importantly, an acceleration of classification and its modernization by making it even more predictive of the genetic and phenotypic relatedness of taxa grouped under the same rank (Konstantinidis and Tiedje, 2007; Rossello-Mora, 2012). It would also be desirable to create a publicly available official website to serve as the platform for the organization and cataloguing of the described diversity and an associated webserver, which will store the DNA sequences and digital protologues and allow external users to query with the data and facilitate further candidate taxa descriptions and research. Efforts such as the Microbial Genome Atlas (available through www.microbial-genomes.org) could serve this goal. We believe that the standards outlined above for classification of uncultivated Bacteria and Archaea could and should be implemented in a few steps, and a committee of experts, supported by an international microbiological society, should be formed in order to govern and supervise the new classification system, in a similar way to ICSP.
Integration with the existing classification of isolated organisms
Our main objective in proposing the abovementioned standards and managing plan was to deviate the least from the current taxonomy of cultivated taxa so that merging of the two systems will be easy in the future. The merging would mostly depend on the implementation of two straightforward changes to the existing code of nomenclature: (i) priority of the names of uncultivated taxa is recognized by the ICNP; and (ii) DNA genome sequence is accepted as the type material for uncultivated taxa. Having a temporary parallel system for uncultured Bacteria and Archaea does not imply a divorce with the bacterial taxonomists. In contrast, it will be of paramount importance that an ‘official’ classification of uncultured taxa does include and is frequently updated with all the hitherto classified taxa. Pluralism does already exist among the different taxonomies for animals and plants (Ereshefsky, 1998). Actually, even within the domain of Bacteria there is already taxonomic pluralism for Cyanobacteria to which some taxonomists apply the International Code of Nomenclature for algae, fungi and plants (Gaget et al., 2015), while the rest apply the ICNP.
It is important to note that, as already recommended for the cultured organisms (Whitman, 2015), using DNA sequence as the type material will expedite descriptions and thus allow the proposed classification system to scale with the increasing number of populations bins and SAGs that become available from environmental surveys. Further, synthetic biological approaches have advanced considerably during the past decade, so that synthesizing individual gene or whole operons to assess—for instance—their function and requirements, probably the most common use of the type material, is possible just from the genome sequence. Finally, it is important to note that the development of a classification to describe yet uncultivated taxa based primarily on genomic sequences is not meant to weaken the efforts of studying principles of biochemistry and microbial physiology on live pure cultures but hopefully to enable it.
References
Abreu F, Martins JL, Silveira TS, Keim CN, de Barros HG, Filho FJ et al. (2007). 'Candidatus Magnetoglobus multicellularis', a multicellular, magnetotactic prokaryote from a hypersaline environment. Int J Syst Evol Microbiol 57: 1318–1322.
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Amann R, Rossello-Mora R . (2016). After all, only millions? MBio 7: e00999-16.
Caro-Quintero A, Konstantinidis KT . (2012). Bacterial species may exist, metagenomics reveal. Environ Microbiol 14: 347–355.
Coleman ML, Chisholm SW . (2010). Ecosystem-specific selection pressures revealed through comparative population genomics. Proc Natl Acad Sci USA 107: 18634–18639.
Collingro A, Toenshoff ER, Taylor MW, Fritsche TR, Wagner M, Horn M . (2005). 'Candidatus Protochlamydia amoebophila', an endosymbiont of Acanthamoeba spp. Int J Syst Evol Microbiol 55: 1863–1866.
Ereshefsky M . (1998). Species pluralism and anti-realism. Philos Sci 65: 103–120.
Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P et al. (2008). The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol 26: 541–547.
Gaget V, Welker M, Rippka R, de Marsac NT . (2015). A polyphasic approach leading to the revision of the genus Planktothrix Cyanobacteria and its type species, P. agardhii, and proposal for integrating the emended valid botanical taxa, as well as three new species, Planktothrix paucivesiculata sp. nov.ICNP, Planktothrix tepida sp. nov.ICNP, and Planktothrix serta sp. nov.ICNP, as genus and species names with nomenclatural standing under the ICNP. Syst Appl Microbiol 38: 141–158.
Glaubitz S, Kiesslich K, Meeske C, Labrenz M, Jurgens K . (2013). SUP05 dominates the Gammaproteobacterial sulfur oxidizer assemblages in pelagic redoxclines of the central Baltic and Black Seas. Appl Environ Microbiol 79: 2767–2776.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM . (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57: 81–91.
Ishoey T, Woyke T, Stepanauskas R, Novotny M, Lasken RS . (2008). Genomic sequencing of single microbial cells from environmental samples. Curr Opin Microbiol 11: 198–204.
Konstantinidis KT, Ramette A, Tiedje JM . (2006). Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl Environ Microbiol 72: 7286–7293.
Konstantinidis KT, Rosselló-Móra R . (2015). Classifying the uncultivated microbial majority: A place for metagenomic data in the Candidatus proposal. Syst Appl Microbiol 38: 223–230.
Konstantinidis KT, Tiedje JM . (2005). Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187: 6258–6264.
Konstantinidis KT, Tiedje JM . (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr Opin Microbiol 10: 504–509.
Lavik G, Stuhrmann T, Bruchert V, Van der Plas A, Mohrholz V, Lam P et al. (2009). Detoxification of sulphidic African shelf waters by blooming chemolithotrophs. Nature 457: 581–584.
Locey KJ, Lennon JT . (2016). Scaling laws predict global microbial diversity. Proc Natl Acad Sci USA 113: 5970–5975.
Luef B, Frischkorn KR, Wrighton KC, Holman HY, Birarda G, Thomas BC et al. (2015). Diverse uncultivated ultra-small bacterial cells in groundwater. Nat Commun 6: 6372.
Luo C, Konstantinidis KT . (2011). Phosphorus-related gene content is similar in Prochlorococcus populations from the North Pacific and North Atlantic Oceans. Proc Natl Acad Sci USA 108: E62–E63 author reply E64–66.
Murray RG, Schleifer KH . (1994). Taxonomic notes: a proposal for recording the properties of putative taxa of procaryotes. Int J Syst Bacteriol 44: 174–176.
Murray RG, Stackebrandt E . (1995). Taxonomic note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int J Syst Bacteriol 45: 186–187.
Oren A . (2016). A plea for linguistic accuracy - also for Candidatus taxa. Int J Syst Evol Microbiol 67: 1085–1094.
Parker CT, Tindall BJ, Garrity GM . (2015). International Code of Nomenclature of Prokaryotes. Int J Syst Evol Microbiol; doi: doi:10.1099/ijsem.0.000778.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW . (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055.
Parte AC . (2014). LPSN—list of prokaryotic names with standing in nomenclature. Nucleic Acids Res 42: D613–D616.
Richter M, Rossello-Mora R . (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131.
Rodriguez RL-M, Konstantinidis KT . (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peer J; doi: doi:10.7287/peerj.preprints.1900v1.
Rodriguez-R LM, Konstantinidis KT . (2014). Bypassing cultivation to identify bacterial species. Microbe Mag.
Rossello-Mora R . (2012). Towards a taxonomy of Bacteria and Archaea based on interactive and cumulative data repositories. Environ Microbiol 14: 318–334.
Rossello-Mora R, Amann R . (2001). The species concept for prokaryotes. FEMS Microbiol Rev 25: 39–67.
Rossello-Mora R, Amann R . (2015). Past and future species definitions for Bacteria and Archaea. Syst Appl Microbiol 38: 209–216.
Rosselló-Móra R, Trujillo ME, Sutcliffe IC . (2017). Introducing a Digital Protologue: a timely move towards a database-driven systematics of Archaea and Bacteria. Syst Appl Microbiol 40: 121–122.
Salman V, Amann R, Girnth AC, Polerecky L, Bailey JV, Hogslund S et al. (2011). A single-cell sequencing approach to the classification of large, vacuolated sulfur bacteria. Syst Appl Microbiol 34: 243–259.
Schloss PD, Girard RA, Martin T, Edwards J, Thrash JC . (2016). Status of the archaeal and bacterial census: an update. MBio 7: e00201-16.
Spencer SJ, Tamminen MV, Preheim SP, Guo MT, Briggs AW, Brito IL et al. (2016). Massively parallel sequencing of single cells by epicPCR links functional genes with phylogenetic markers. ISME J 10: 427–436.
Stackebrandt E, Ebers J . (2006). Taxonomic parameter revisited: tarnished gold standards. Microbiol Today 33: 152–155.
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kampfer P, Maiden MC et al. (2002). Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52: 1043–1047.
Stackebrandt E, Goebel BM . (1994). Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in Bacteriology. Int J Syst Bacteriol 44: 846–849.
Sutcliffe IC . (2015). Challenging the anthropocentric emphasis on phenotypic testing in prokaryotic species descriptions: rip it up and start again. Front Genet 6: 218.
Whitman WB . (2015). Genome sequences as the type material for taxonomic descriptions of prokaryotes. Syst Appl Microbiol 38: 217–222.
Yarza P, Yilmaz P, Pruesse E, Glockner FO, Ludwig W, Schleifer KH et al. (2014). Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12: 635–645.
Acknowledgements
Our work is supported by US NSF (awards 1241046 and 1356288 to KTK) and the Max Planck Society (to RA). RR-M acknowledges the support by the Spanish Ministry of Economy and Competitiveness project CLG2015_66686-C3-1-P, also supported with European Regional Development Fund (FEDER) funds. We also thank Aharon Oren, Nikos Kyrpides and several of the participants of the ISME16 roundtable Candidatus for helpful discussions related to the classification of the uncultivated majority. Jim Tiedje and Barny Whitman are particularly acknowledged for their helpful discussions related to the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Konstantinidis, K., Rosselló-Móra, R. & Amann, R. Uncultivated microbes in need of their own taxonomy. ISME J 11, 2399–2406 (2017). https://doi.org/10.1038/ismej.2017.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.113
This article is cited by
-
Unraveling the phylogenomic diversity of Methanomassiliicoccales and implications for mitigating ruminant methane emissions
Genome Biology (2024)
-
An in-depth evaluation of metagenomic classifiers for soil microbiomes
Environmental Microbiome (2024)
-
An abundant bacterial phylum with nitrite-oxidizing potential in oligotrophic marine sediments
Communications Biology (2024)
-
Freshwater genome-reduced bacteria exhibit pervasive episodes of adaptive stasis
Nature Communications (2024)
-
A novel plant growth-promoting rhizobacterium, Rhizosphaericola mali gen. nov., sp. nov., isolated from healthy apple tree soil
Scientific Reports (2024)
