
Abstract
Water from The Cedars springs that discharge from serpentinized ultramafic rocks feature highly basic (pH=~12), highly reducing (Eh<−550 mV) conditions with low ionic concentrations. These conditions make the springs exceptionally challenging for life. Here, we report the metagenomic data and recovered draft genomes from two different springs, GPS1 and BS5. GPS1, which was fed solely by a deep groundwater source within the serpentinizing system, was dominated by several bacterial taxa from the phyla OD1 (‘Parcubacteria’) and Chloroflexi. Members of the GPS1 community had, for the most part, the smallest genomes reported for their respective taxa, and encoded only archaeal (A-type) ATP synthases or no ATP synthases at all. Furthermore, none of the members encoded respiration-related genes and some of the members also did not encode key biosynthesis-related genes. In contrast, BS5, fed by shallow water, appears to have a community driven by hydrogen metabolism and was dominated by a diverse group of Proteobacteria similar to those seen in many terrestrial serpentinization sites. Our findings indicated that the harsh ultrabasic geological setting supported unexpectedly diverse microbial metabolic strategies and that the deep-water-fed springs supported a community that was remarkable in its unusual metagenomic and genomic constitution.
Similar content being viewed by others


Introduction
Serpentinization is a process whereby water reacts with ultramafic mantle rock (peridotite) to produce a new suite of minerals (including serpentine), ultrabasic (pH≈12) fluid, molecular hydrogen and, under some conditions, reduced carbon in the form of methane (Sleep et al., 2004; Etiope and Lollar, 2013; Morrill et al., 2013; Schrenk et al., 2013). Such serpentinization settings are thought to have been common on early Earth, with both hydrogen and methane formed through reactions associated with serpentinization. Therefore, serpentinizing environments have been implicated as locations that may have supported early microbial life (Schulte et al., 2006; Sleep et al., 2011).
The Cedars is an active serpentinization site located in northern California (Barnes et al., 1967) with spring waters that have extremely high pH (~12), very low Eh (~−500 mV or less) and are rich in Ca2+ (~1 mM), hydrogen and methane gas, and contain very low levels of dissolved organic carbon, total inorganic carbon, ammonium, phosphate and electron acceptors (oxygen, nitrate, sulfate) (Morrill et al., 2013; Kohl et al., 2016). While these properties describe an environment that should be exceptionally challenging for microbial life, our previous studies have revealed a diverse microbial community associated with these ultrabasic springs (Suzuki et al., 2013), several of the members of which are now in culture (Suzuki et al., 2014).
Geochemical studies of two neighboring springs at The Cedars have indicated that they are fed by different groundwater sources flowing through the serpentinizing system (Figure 1): (1) GPS1, being fed by a deep groundwater source that interacts with both the peridotite body and the kilometer-deep marine sediments of the Franciscan Subduction Complex, and, (2) BS5, fed by a mixture of ~15% of the deep groundwater source and 85% of a shallow groundwater source that interacts only with the overlying peridotite body (Morrill et al., 2013).

A conceptual representation of the proposed water sources at The Cedars Allows show the direction of water flow. Numbers in percentage indicate the fraction of deep or shallow source at each spring.
Microbial analyses of the springs have revealed that the two spring water sources (deep and shallow) support very different microbial communities (Suzuki et al., 2013). The BS5 shallow community was similar to communities seen in other terrestrial serpentinization sites, being dominated by Proteobacteria (Brazelton et al., 2012, 2013; Tiago and Verissimo, 2013; Quemeneur et al., 2015). Several strains of the Proteobacteria have already been isolated from the BS5. These strains are members of a proposed genus Serpentinomonas that is closely related to the genus Hydrogenophaga in the Betaproteobacteria (Suzuki et al., 2014). Serpentinomonas is a dominant microbial community member in almost every terrestrial serpentinization site that has been reported (Brazelton et al., 2012, 2013; Tiago and Verissimo, 2013; Quemeneur et al., 2015). Physiological, genomic and transcriptomic studies of the Serpentinomonas strains have indicated that they use a hydrogen-oxidizing, oxygen-respiring, autotrophic metabolism (Suzuki et al., 2014). In marked contrast, the deep community of GPS1 was populated mostly by uncharacterized bacterial phyla or families, as well as one archaeal taxon (Suzuki et al., 2013). Phylotypes similar to some of those seen in the deep groundwater source have been reported from an alkaline hydrothermal field in New Caledonia and an oceanic serpentinization site, Lost City (Brazelton et al., 2010; Quéméneur et al., 2014; Postec et al., 2015).
Geochemical features and microbial taxonomic diversities of various serpentinization sites have been described previously (Brazelton et al., 2010, 2013; Tiago and Verissimo, 2013; Quéméneur et al., 2014, 2015; Sanchez-Murillo et al., 2014; Postec et al., 2015; Miller et al., 2016). In addition, several metagenomic studies of serpentinization sites have further described the presence of genes related to the methane and hydrogen cycling in the serpentinizing systems (Brazelton and Baross, 2009; Brazelton et al., 2012, 2016). However, life strategies used by the majority of microbial community members living in the serpentinization systems remain elusive, especially for the members associated with the deep subsurface serpentinization.
To illustrate the life strategies of the individual microorganisms associated with the serpentinization occurring at The Cedars, we used a genome-centric metagenomic approach and report here the genomic features and metabolic capabilities with a special focus on the dominant members from each groundwater source (that is, shallow and deep). With regard to the members in the shallow groundwater source of BS5, this study complemented and extended our previous physiological studies of the Serpentinomonas strains (Suzuki et al., 2014), as well as other ‘familiar’ Proteobacteria that have the capability of a hydrogen-based autotrophic metabolism. In contrast, the GPS1 community was markedly different, with the dominant members represented by a group of genomes in the candidate phylum OD1 (‘Parcubacteria’) (Wrighton et al., 2012; Rinke et al., 2013; Brown et al., 2015), and two genomes in the phylum Chloroflexi. The OD1 genomes were enigmatic, being very small, and lacking a number of genes needed for independent life, including: (1) ATP synthase (ATPase); (2) early-stage glycolysis genes; and (3) key biosynthetic genes. Metabolic reconstruction from any single OD1 draft genome failed to reveal known metabolic strategies. Finally, while nearly all the members in the GPS1 community were in the domain Bacteria, no bacterial type (F-type) ATPase genes were seen, either in the binned genomes or in the metagenomes of GPS1.
Materials and methods
Sampling sites and sample collection
Samples for microbiological analysis were collected from two different ultrabasic springs (Morrill et al., 2013; Suzuki et al., 2013; Kohl et al., 2016), GPS1 (elevation 273 m, N: 38°37.268′, W: 123°8.014′) and BS5 (elevation 282 m, N: 38°37.282′, W: 123°7.987′) at two different sampling times, September in 2011 and June in 2012. Each year, water samples (1000 L from GSP1, and 200 L from BS5) for the metagenomic analysis were retrieved using a peristaltic pump. The cells in the water were collected on 0.22 μm filters (Millipore, Billerica, MA, USA) using PFA in-line filter holders (Advantec, Dublin, CA, USA) attached to Tygon Chemical Tubing (Masterflex, Gelsenkirchen, Germany) or Chem-Durance Tubing (Masterflex). The filter for the microbial cell collection was replaced every several hours. The filtered cells were immediately frozen with dry ice at each sampling site and kept at the temperature of dry ice during the transportation. The samples were stored at −80 °C until the DNA was extracted.
Water samples collected for tritium age dating were stored with no headspace in 500 ml Nalgene bottles. Tritium concentrations were determined using the electrolytic tritium (E3H) method. Analyses were performed at Isotope Tracer Technologies Inc. in Waterloo, Ontario. The enrichment technique passes an electrical current through the water to isolate the tritium and deuterium molecules while breaking down water into its constituents of hydrogen and oxygen. Tritium was measured using liquid scintillation counting.
Sequencing and sequence assembly
A MoBio PowerBiofilm RNA Isolation Kit (MoBio, Carlsbad, CA, USA) was used to achieve the highest yield of total nucleic acids. To extract total nucleic acids, the standard MoBio RNA extraction method was followed with some modifications (Ishii et al., 2013). Briefly, the filter, having the collected cells, was directly added to the power beads containing Solution BFR1/β-mercaptoethanol. After the inhibitor removal steps, the DNA removal step was eliminated and the process was directly moved to the sample washing step. Total nucleic acid was then eluted in nuclease-free water. Extracted total nucleic acid was separated using the AllPrep DNA/RNA Mini Kit (Qiagen, Germantown, MD, USA) (Ishii et al., 2013). The prepared DNAs were sequenced using the HiSeq2000 (Illumina, San Diego, CA, USA) using the 101 bp read length option with paired-end (~250 bp insertion size) library. The paired-end libraries were prepared as described previously (Suzuki et al., 2014).
The de novo assembly of the metagenomic sequences into contigs by scaffolding was conducted using CLC de novo Assembly Cell (version 4.0; CLC Bio, Cambridge, MA, USA) with a bubble size of 800 bp and a kmer length of 33 bp for GPS1 metagenomes, and a bubble size of 800 bp and a kmer length of 43 bp for BS5 metagenomes. Only contigs over 500 bp were supplied for the following analyses as similar to the previous study (Ishii et al., 2013). The mean coverage and the number of mapped raw reads for each contig were determined during the assembly step in CLC Genomics Workbench (version 6.0.3; CLC Bio). The mean coverage of contigs was used as the relative frequency within the community. Contigs were processed with the JCVI prokaryotic metagenomic open reading frame (ORF) calling and annotation pipeline and taxonomically assigned based on the most abundant taxonomic information of the peptides in the specific contig as described previously (Ishii et al., 2013). Metagenome sequences from this study have been deposited in DDBJ BioProject under accession number PRJDB2971 and the assembled contigs have been deposited in DDBJ/EMBL/GenBank under accession numbers BBPD01000001–BBPD01034182, BBPE01000001–BBPE01037294, BBPF01000001–BBPF01025471 and BBPG01000001–BBPG01016475.
Genome binning
Bin-genomes of the dominant microbial community members within The Cedars springs were initially extracted by grouping the contigs using four criteria: (i) mean coverage, (ii) G+C content, (iii) predicted taxonomy and (iv) length of the contigs as described in the previous study (Ishii et al., 2013). In addition, cross-mapping plots between mean coverage of contigs from different metagenomic sequence samples were also used for refining the bin-genomes (Albertsen et al., 2013). The cross-read mapping analyses were run using Map Reads to reference algorithm in CLC Genomics Workbench (version 6.5) with the following settings: 0.7 of minimum length and 0.97 of minimum similarity fractions. The parameters were selected to increase % read mapped to contigs and also to reduce % read mapped to multiple contigs. Four assemblies from different springs and years were individually provided as a reference for the raw reads mapping of four metagenomic sequences (only single side of paired-end sequencing). Subsequent to the contig frequency-based clustering, the bin-genomes were further refined by using their tetranucleotide frequency pattern (Dick et al., 2009; Albertsen et al., 2013; Ishii et al., 2013).
For further bin-genome taxonomic clustering, all bin-genomes were compared in a pairwise manner based on average nucleotide identity (ANI) and correlation of the tetranucleotide signatures (TETRA) with the default parameters selected in the software JSpecies V1.2.1 (Richter and Rossello-Mora, 2009). A group of bin-genomes showing both ANIb values over 97% and TETRA correlation values more than 0.99 was defined as the same taxonomic group having species/strain level similarity, and named operational candidate species (OCS) in this study. A representative genome from the OCS was then chosen based on: (1) the most complete compliment of single-copy genes, (2) the highest total bin length and (3) the lowest scaffold fragmentation.
A BLAST Ring image generator was used for visualizing a genome as a circular image for comparison of multiple The Cedars bin-genomes from different springs and years (Alikhan et al., 2011).
Functional annotation
Contigs associated with the bin-genomes were processed in the Microbial Genome Annotation Pipeline (http://www.migap.org/) for further ORF calling and functional annotation (Sugawara et al., 2009). In the pipeline, ORFs were identified from the contigs by MetaGene Annotator, and then the predicted ORFs were used to search against reference databases including RefSeq, TrEMBL and COG. For the KEGG orthology (KO) assignment for each ORF, we used the KEGG Automatic Annotation Server with the SBH (single-directional best hit) method set to 37 as the threshold assignment score (Moriya et al., 2007). Microbial cell activity- and metabolism-associated marker genes were selected from the KEGG module or KEGG pathway databases and analyzed as described previously (Ishii et al., 2015). The completeness of KEGG modules for each bin-genome was also analyzed by the Metabolic And Physiological Potential Evaluator web server (Takami et al., 2016).
Estimation of genome completeness, measurement of replication rates and analysis of genome size distribution
The members of the candidate phylum OD1 are known to have reduced metabolic potentials; therefore, all the high-coverage draft genomes of OD1 organisms (RAAC4 (Kantor et al., 2013), ACD1, ACD7, ACD11, ACD18, ACD81 (Albertsen et al., 2013)) do not code all of the 107 KOs that were proposed as single-copy universal marker genes for all bacterial genomes (Dupont et al., 2012; Ishii et al., 2013). Consequently, the genes lacked by all of the high-coverage OD1 genomes were excluded from the list of 107 KOs for the genome completeness analysis. Ultimately, 78 single-copy KOs remained for bacterial genome completeness estimation (Supplementary Data 1). All the 43 universal marker genes that were previously determined to be appropriate for the completeness analysis of Candidate Phylum Radiation (CPR) genomes (Brown et al., 2015) were included in the 78 single-copy genes that we selected. The completeness of archaeal bin-genomes was assessed by using 137 marker genes for Archaea (Ishii et al., 2013) (Supplementary Data 2). The marker gene list was constructed based on the comparison of 99 archaeal genomes using a method described previously (Kawai et al., 2011).
Genome replication rate was estimated by index of replication (iRep) (Brown et al., 2016). After the metagenomic raw read was mapped to the individual bin-genome by using Burrows-Wheeler Alignment tool with the default parameters selected (Li and Durbin, 2009), an iRep was calculated based on the sequencing coverage trend that resulted from bidirectional genome replication from a single origin of replication (Brown et al., 2016).
To examine the genome size distribution of The Cedars organisms among each taxon, genome size information for all bacterial and archaeal organisms within the NCBI genome database (total of 2900 genomes with complete status as of 6 March 2014) were used as references. Regarding OD1, recovered OD1 draft genomes (Albertsen et al., 2013; Kantor et al., 2013; Rinke et al., 2013; Brown et al., 2015; Anantharaman et al., 2016) with over 85% of the completeness were used for the genome size assessment.
Phylogenetic analysis of the OD1 draft genomes
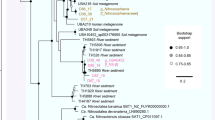
Phylogenetic analysis was carried out using a protein marker ribosomal protein S3. The reference ribosomal protein S3 protein sequences associated with the phylum were downloaded from the NCBI database. Clustering was based on the reports from Brown et al. (2015) and Anantharaman et al. (2016).
ATPase distributions among various taxa and phylogenetic analyses of the NtpA
To determine how the genes encoding F- and A(V)-type ATPase (A-ATPase) were distributed in the sequenced genomes of various taxa, we examined the distribution for all of the organisms in the KEGG database (March 2014): 2978 total organisms, including 228 Eukaryotes, 2585 Bacteria and 165 Archaea. The targets for the analysis were the genes encoding subunit A of ATPases: atpA gene for the F-type ATPase (F-ATPase) and ntpA gene for the A-ATPase.
To determine the phylogenetic distribution of A-ATPase of The Cedars organisms, the reference sequences were collected from a KO database. Protein ID lists for the amino-acid sequences of NtpA (K02117) were obtained from a KO database and 577 amino-acid sequences of NtpA were retrieved from the Uniplot database (March 2014). Amino-acid sequences annotated as NtpA by KEGG Automatic Annotation Server in The Cedars metagenomes were aligned with those retrieved from the database. Sequence alignment was performed with MUSCLE (Edgar, 2004) and the trees were created by using maximum likelihood with RaxML (Stamatakis et al., 2008).
For further examination of the closest relatives of each NtpA of The Cedars organisms, we ran blast against the nr (non-redundant protein sequences) database (NCBI) and collected the 20 closest relatives of those sequences as the references. The methods for the alignment and tree constructions were the same described above.
Microscopic observation
Cell samples for catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) analysis were collected at GPS1 in June 2013. Owing to the low cell density, concentration of the cells was required for the effective detection of OD1 organisms. Cells were concentrated with a tangential flow filtration system (Millipore). This system was used instead of an in-line filtration system because of the reduced pressure experienced by the cells. The membrane pore size for the filtration was 0.1 μm. After concentration, cells were collected onto a polycarbonate membrane filter and fixed with 4% paraformaldehyde in CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) buffer solution. The CAPS buffer was adjusted to pH 11 with sodium hydroxide. The fixation procedure was described previously (Hoshino et al., 2008). Microbial community compositions determined from the cell samples collected with two different systems, that is, tangential flow filtration and in-line filter, were nearly identical. The determination of community composition was carried out as described previously (Suzuki et al., 2013).
Using the nearly full-length of 16S rRNA sequences obtained from GPS1 (Suzuki et al., 2013), an oligonucleotide probe was designed using the ARB software (Ludwig et al., 2004) to specifically detect OD1 cells, and subsequently evaluated for its specificity using clone-FISH (Schramm et al., 2002). Using the optimized conditions, CARD-FISH was performed to detect OD1 cells concentrated onto the polycarbonate membrane filter as described by Hoshino et al. (2008). Briefly, the filter was cut into small sections and put in 0.2 ml PCR tubes. After permeabilization with lysozyme, the filter sections were hybridized at 37 °C for 2 h, followed by signal amplification with Alexa488-labeled tyramide. The cells were visualized under a BX51 epifluorescence microscope (Olympus, Tokyo, Japan). The nonEUB388 probe (the reverse complement of EUB388) was used as a negative control for the CARD-FISH to characterize nonspecific binding fluorescence. Areas of interest (containing fluorescent cells) were marked by laser microdissection (CellCut Plus: Molecular Machines and Industries, Eching, Germany), and the surfaces of those filter sections were coated by carbon. Subsequently, scanning electron microscopy energy dispersive X-ray spectroscopy analysis was conducted to determine the elemental composition of mineral particles harboring OD1 cells using scanning electron microscope (SU1510; Hitachi High-Tech, Tokyo, Japan).
Results
Genome recovery from metagenomic sequences of The Cedars springs
Our previous work had revealed that the microbial communities of BS5 and GPS1 were stable over time, very different from each other depending on the groundwater source (deep versus shallow) (Suzuki et al., 2013). The geochemical properties of BS5 and GPS1 have remained stable over a 7-year period (Morrill et al., 2013), and the average values over multiple sampling periods are shown in Supplementary Table 1. To gain further insights into the biological differences between these two springs, we conducted four deep metagenomic sequence determinations in two different years (2011 and 2012) from these two springs, with the goal of characterizing the dominant microbial groups and the metabolic potential of both the individuals and the communities.
After assembling the raw sequence reads into contigs (Supplementary Table 2), the contigs were binned for recovering draft genomes (bin-genomes) using genome-binning approaches described previously (Albertsen et al., 2013; Ishii et al., 2013). Briefly, the contigs from each metagenomic sequence set were clustered to recover bin-genomes, respectively, by using the differences in DNA frequency levels (mean coverage), taxonomic assignments, length and GC contents (Figures 2a and c and Supplementary Figures 1A and D). While the method of clustering assembled contigs with multiple criteria retrieved bin-genomes for most of the organisms (Figure 2), the bin-genomes for some of the major organisms turned out to be a mixture of genomes from multiple organisms (for example, unc4B and unc5 (Figure 2a), unc4B and unc17 (Figure 2c) and bet2 and bet4 (Figure 2c)). Therefore, to improve the quality of bin-genomes, differences of mean coverage for each contig between GPS1 and BS5 and/or between 2011 and 2012 were used for further clustering (Figures 2b and d and Supplementary Figures 1B and D) in combination with tetranucleotide frequencies (Dick et al., 2009). These strategies enabled the recovery of bin-genomes that could not be clustered in the initial genome-binning effort, and the linkage of each bin-genome between the samples from different springs and years were also accurately identified. Using this approach, 74 bacterial and five archaeal bin-genomes were successfully recovered from the contigs delivered from four sets of metagenomic sequences (Supplementary Data 3). Between 86.6 and 91.9% of the entire raw reads from each metagenomic sequence were mapped back to the assembled contigs, while 68.0–81.6% of the raw reads were mapped to the recovered bin-genomes (Supplementary Table 2), indicating that almost all of the genomic information from the respective communities was included.

Genome clustering from the assembled contigs of metagenomic sequences of The Cedars springs GPS1 and BS5. Samples collected from GPS1 and BS5 in 2011 are shown as GPS11 or BS5B11, respectively. Colored circles indicate clusters of individual bin-genomes. Bin-genomes were identified using the estimated taxonomic classification (color of dots), length (size of dots), GC content and mean coverage of contigs. Colors for the circles denote groundwater sources where the bin-genomes are associated, that is, red circles show the deep members, while blue circles show the shallow member. Numbers/letters shown near the circles correspond to the bin-genome IDs in (a). (a and c) Clustering based on the mean coverage versus GC% plot. (b) Clustering for the members of deep groundwater based on coverage differences between the two springs. (d) Clustering for the members of shallow groundwater based on the coverage differences of 2 years’ metagenomes.
Cross-mapping of the 79 bin-genomes indicated the presence of ‘nearly identical bin-genomes’ recovered from different metagenomic data sets. For the further grouping of the members affiliated with the same taxonomic group, the bin-genomes were compared in a pairwise manner based on ANI and correlation of the TETRA (Richter and Rossello-Mora, 2009). Since it was previously reported that a 95–96% ANI threshold can be readily used as an objective boundary for species circumscription, especially when it is reinforced by high TETRA correlation values, groups of bin-genomes that showed high ANIb values (over 97%) and high TETRA correlation values (over 0.99) were defined as a same population having species/strain level similarity. In this study, the taxonomic group of population was defined as OCS (Supplementary Data 4). These efforts led to the identification of 27 OCSs in the whole metagenomic data set, and 16 of those were related to the deep groundwater source (Supplementary Figure 2) and 11 of those were related to the shallow groundwater source. A representative genome from each OCS was then chosen based on the most complete complement of single-copy housekeeping gene families, highest total bin-genome length and lowest scaffold fragmentation (Supplementary Data 3 and Figure 3a). Genome completeness analysis of the representative genomes from each OCS revealed that 17 bin-genomes had >90% completeness (Figure 3a), as estimated by the presence of 78 bacterial and 136 archaeal single-copy housekeeping genes (Supplementary Data 1 and 2) (Dupont et al., 2012).

Overview of genome recovery. (a) Genomic features of representative bin-genomes of OCS. Completeness was estimated as fraction of the number of identified and the number of expected core genes within the phylogenies. Values of the iRep infer microbial population replication rates. *The number shows unfiltered iRep value. (b and c) Relative frequencies of each OCS within GPS11 (b) and BS5B11 (c) estimated from a relative abundance of 16S rRNA gene (16S analysis), percentage of mapped raw reads to each bin-genome (Raw reads), an average value of reads per kilobase per million mapped reads (RPKMs) of each bin-genome (Coverage), an average RPKM value of 12 single-copied housekeeping genes (Core genes) and an RPKM value of 12 individual single-copied housekeeping genes.
Community composition of GPS1 and BS5 estimated from relative abundance of single-copy genes
The relative frequencies of the dominant OCSs were evaluated by the abundance of single-copy housekeeping genes encoded by the respective bin-genomes (Wu and Eisen, 2008), and showed that the 27 OCSs accounted for over 97% of the community for each sample (Figures 3b and c and Supplementary Figures 1C and F). The phyla with which the most abundant OCSs in GPS1 were affiliated were the candidate phylum OD1 (unc1, unc4A, unc4B, unc5 and unc17), followed by the phylum Chloroflexi (chl2 and chl3). OCSs belonging to the two taxa, that is, phyla OD1 and Chloroflexi, consistently accounted for 90% of the GPS1 community in the 2-year study (Figure 3b). Meanwhile, the dominant populations in BS5 belonged to the Alpha-, Beta- and Gammaproteobacteria, which constituted ~50% of the community (Figure 3c).
The phylogenetic analysis based on the 16S rRNA gene for The Cedars communities have been reported previously (Suzuki et al., 2013) and the operational taxonomic unit ID and the corresponding OCSs have been linked and shown in the column of operational taxonomic unit ID in Figure 3a. Since comprehensive clustering of CPR bacteria including OD1 has been recently reported by Brown et al. (2015) and Anantharaman et al. (2016), phylogenetic analysis was carried out using a protein marker ribosomal protein S3, which has been independently used as a phylogenetic marker because of its strong phylogenetic signal. The result showed that bin-genome IDs unc1, unc4A, unc4B and unc17 were placed within a phylogenetic group of ‘Candidatus Nealsonbacteria’, which was proposed by Anantharaman et al. (2016), while the bin-genome unc5 did not cluster with any sequenced draft genomes (Supplementary Figure 3).
Metabolic reconstruction of The Cedars dominant microorganisms
Hydrogen is an abundant energy source in all of The Cedars springs (Morrill et al., 2013). However, given that both the dissolved organic carbon and the well-known electron acceptors for microorganisms (oxygen, nitrate and sulfate) were very low or below detectable levels in all of the analyzed serpentinizing springs at The Cedars (Morrill et al., 2013), both microbial fermentation and respiration were assumed to be minor processes in the springs (Supplementary Table 1). To understand the life strategies taken by the microbial communities in The Cedars ultrabasic springs, metabolism-related pathways of each organism were reconstructed based on the gene contents of the bin-genomes from the two sources. The analysis was performed in the KEGG module and detailed data sets for all the bin-genomes are shown in Supplementary Data 9 and 10. In addition, Metabolic And Physiological Potential Evaluator analysis was performed for the dominant representative bin-genomes of each OCS to identify the module completion ratio. The results are shown in Supplementary Data 11 and summarized in Table 1.
Predicted metabolic strategies of the shallow groundwater community
Representative bin-genomes of each OCS from the metagenome of BS5 showed that the dominant bin-genomes associated with the shallow groundwater belong to the Alpha-, Beta-, Gamma- and Deltaproteobacteria. Metabolic reconstructions of those revealed that the gene sets encoding various hydrogenases were common, and all the bin-genomes associated with the shallow groundwater had the genes indicative of respiratory metabolism; that is, genes for NADH: quinone oxidoreductase, the TCA cycle and terminal electron acceptor reductases (for example, cytochrome c oxidase, nitrate reductase, sulfate reductase) (Table 1 and Supplementary Data 10). It appeared that the Proteobacterial members in the shallow groundwater were capable of respiratory metabolism, presumably coupled to hydrogen oxidation.
Predicted metabolic strategies of the deep groundwater community
Examination of the metagenome of GPS1 provided a very different picture; only a few partial respiration-related gene sets were observed, and not a single example of an entire respiratory pathway was detected (Table 1 and Supplementary Data 10). This was verified as expected with examination of the bin-genomes of the GPS1 OCSs, where virtually no respiration-related genes were present. Unless some unknown or hypothetical genes are capable of catalyzing these functions, it must be concluded that GPS1 harbors a community incapable of respiratory metabolism.
As for fermentation, OCSs affiliated with the phylum OD1 (unc1, unc4A, unc4B, unc5 and unc17), which was the most dominant taxon in GPS1, lacked the genes responsible for several of the initial steps of the glycolysis: those involved with the conversion of glucose to glyceraldehyde-3-phosphate (Supplementary Data 10 and 11), suggesting that at least the dominant OD1 organisms in GPS1 were incapable of gaining energy via sugar fermentation.
The bin-genomes of the other members of the deep community members (chl2, chl3, unc8, fir7, fir9 and fir15) also lacked the full complement of glycolysis, suggesting that sugar fermentation may not be an important part of their energy metabolism, consistent with the chemical properties of the deep source (high hydrogen, very low dissolved organic carbon and very low or no sugars).
A potential metabolism based on the analysis of representative bin-genomes in the phylum Chloroflexi (chl2 and chl3), which was the second most dominant taxon in the community, is that of acetogenesis. Both chl2 and chl3 have gene sets for the Wood–Ljungdahl pathway and hydrogenases (mvhA,D,G and nuoE,F,G) (Ragsdale and Pierce, 2008), indicating that they could be acetogens, gaining energy via acetate production from hydrogen and inorganic carbon, although the bicarbonate concentration was very low in the springs (Table 1 and Supplementary Data 10 and 11).
Predicted biosynthetic metabolism(s)
As summarized in Table 1, the two most dominant OD1 bin-genomes related to the deep groundwater, unc1 and unc4A, lacked almost all known genes that are required for the de novo biosynthesis of nucleotides, amino acids, fatty acids or lipids, indicating a severe biosynthetic limitation (Supplementary Data 11). Although the OD1 genomes in The Cedars had similar percentage of unpredicted genes per genome to those of the other organisms there (percentage of predicted genes: OD1=73–77%; others=60–80%) (Supplementary Data 14), it could not be excluded that some of these unidentified genes could be related to biosynthesis or energy metabolisms of OD1s. The OD1 bin-genomes did, however, encode the modules required for replication, transcription and translation, and also synthesis of peptidoglycan and pili (Supplementary Data 9). As is often seen in this phylum, introns were found in the 16S rRNA genes (Brown et al., 2015) (Supplementary Figure 4): the 16S rRNA genes of unc4B and unc5 contained various long internal introns, and that of the unc1 contained two introns at the edge.
In contrast, the examination of the bin-genomes of chl2 and chl3 indicated that they should be capable of synthesizing biomolecules required for independent life and for performing replication, transcription and translation (Table 1 and Supplementary Data 9 and 10). All members in the shallow groundwater encoded biosynthesis pathways required for independent life.
Genome sizes
Comparison of the genome sizes for the high-coverage representative bin-genomes (over 85% of completeness) revealed that genome size of the microbial members associated with the deep-water source (candidate phylum OD1, other candidate phyla, Chloroflexi, Firmicutes or Euryarchaeota) were the smallest genomes reported for each of these taxa (Figures 4a–c and Supplementary Figures 5A and E). Although it is unlikely based on estimates of genome completion, it could not be ruled out that some of the very small genomes are as yet incomplete. The sizes of bin-genomes from the shallow water source, which were assigned to Proteobacteria, varied widely, ranging from slightly smaller than those of similar taxa from other environments, to nearly the smallest sizes within the genomes of a given taxon (Supplementary Figures 5A–C).

GC content versus genome size for the bin-genomes of The Cedars members with reference genome sequences in each taxonomic group. Red and black dots denote the bin-genomes from The Cedars organisms or genomes in NCBI genome respectively. Genome size and GC contents information for the references were obtained from all completed bacterial genomes in the NCBI genome database and also recovered high-coverage draft genome sequences affiliated with a candidate phylum OD1 (Albertsen et al., 2013; Kantor et al., 2013; Brown et al., 2015; Anantharaman et al., 2016) (over 85% of completeness).
ATPase distribution
ATP synthases (ATPases) are key enzymes that mediate ATP production from the proton motive force at the cell membrane. ATPases are composed of a highly conserved family of proteins that tend to be ‘domain specific’: F-ATPase for bacteria and eukaryotes (von Ballmoos et al., 2008), and A-ATPase for archaea and some bacteria (Muller and Gruber, 2003). The few known exceptions lend support to the notion that these enzymes are ancient, and reasonably easy to trace, with regard to their origin (Hilario and Gogarten, 1993; Koonin and Martin, 2005; Mulkidjanian et al., 2009).
While the GPS1 community was dominated by bacterial taxa, not a single gene sequence indicative of an F-ATPase gene cassette was seen in either the metagenome or any of the bin-genomes, while abundant sequences encoding A-ATPases were documented (Table 1 and Supplementary Data 9). Phylogenetic analysis of amino-acid sequences of the NtpA (A subunit of A-ATPase) indicated that NtpA in chl2 (phylum Chloroflexi) was in the phylum Crenarchaeota (Supplementary Figure 6), although no OCSs of this phylum were detected in The Cedars metagenome (the detection limit was 0.1%). Further detailed phylogenetic analysis revealed that the NtpA in chl2 was closely related to a sequence from a bin-genome of an uncultured Chloroflexi bacterium (RBG-2) recovered from a uranium-contaminated aquifer at The Rifle Integrated Field Research Challenge (Supplementary Figure 7). NtpA from the remaining GPS1 bacterial members were clustered into a diverse clade of bacterial A-ATPase proteins. NtpA from several candidate phyla (bin-genomes unc4A, unc6, unc8, unc11) were found to be deeply branched and closely related to the NtpA from thermophiles within phyla Dictyoglomi and Caldiserica, while those from the phylum Firmicutes (bin-genomes fir7, fir9, fir14, fir15) were clustered with NtpA from alkaliphilic Dethiobacter alkaliphilus (Supplementary Figure 7) (Sorokin et al., 2008). In addition, four of the five OD1 bin-genomes have no recognizable ATPase genes, even though the presence of ATP-dependent transporters and various kinase genes on the genomes is consistent with ATP being an important metabolite for these microbes. Meanwhile, the ATP synthase distribution in the shallow groundwater bin-genomes was ‘as expected’ (all bacterial genomes had only the F-type (bacterial type) ATPase genes) (Table 1), which may indicate that the unusual distribution documented in the deep groundwater community is not attributed to life at high pH.
Replication rate
Although the majority of the bin-genomes affiliated with the phylum OD1 lacked genes or pathways responsible for the biogenesis, ATP synthesis and energy metabolisms, the bin-genomes accounted for 62–72% of the GPS1 community over the two consecutive years, indicating that genome replication of the OD1 members must be occurring in the GPS1 community. To measure replication rates of the community members, the iRep algorithm developed by Brown et al. (2016) was applied to the bin-genomes recovered from the four different metagenomic data sets (Figure 3a and Supplementary Data 3). Median of the iRep values of OD1 bin-genomes ranged between 1.39–1.43, which was similar to the median iRep values of the whole bin-genomes constituting the community (whole bin-genomes=1.40–1.45) (Supplementary Figure 8). Brown et al. (2016) reported that median iRep values from uncultivated, groundwater-associated CPR bacteria were significantly lower (CPR=1.34) compared with those from premature infant microbiomes (infant=1.42); thus, the CPR bacteria only rarely replicated quickly in subsurface communities undergoing substantial changes in geochemistry. However, the microbial members in The Cedars appeared to act differently, with the replication rate of members in the phylum OD1 being similar to those in the other phyla in The Cedars springs.
Environmental variables between the two ultrabasic spring sources
Although the shallow and deep groundwater sources shared many geochemical features such as high pH and low Eh, the two springs hosted very different microbial communities with regard to the taxa and their predicted metabolic capabilities. While it has been already identified that the sodium, chloride and bromine are significantly higher in the deep groundwater, to further identify the environmental factors that distinguish the two sources, additional biogeochemical analyses have been performed.
Radioactive 3H, with a half-life of 12.43 years, in spring water provides a relative age of groundwater based on when the water was last in contact with the atmosphere. The 3H concentration values determined in this study revealed that the two groundwater sources (that is, shallow and deep) were different in their ages, that is, the water discharging out of GPS1 was submodern (<0.8 Tritium Unit (TU)) originated before the 1950s, while the water from the BS5 was a mixture of modern and submodern waters (2.3 TU). As it was previously determined that these groundwaters were of meteoric origin (Morrill et al., 2013), these results indicated that the deep groundwater would have longer flow paths and residence time in the subsurface, and therefore would be older, compared with springs fed by a mixture of deep and shallow groundwater.
Interactions of microbes with minerals
At The Cedars, calcium carbonate naturally precipitates when the spring water comes into contact with atmospheric air. Physiological studies of Serpentinomonas strains, which are the dominant Betaproteobacteria in the shallow groundwater, revealed that the isolates could fix CO2 from calcium carbonate and dissolve solid calcium carbonate as the cell population increased (Suzuki et al., 2014). Thus, interaction of Serpentinomonas strains and calcium carbonate must be occurring in the spring.
To visualize the localization of the dominant organisms in the GPS1, OD1 organisms were detected with CARD-FISH analysis (Hoshino et al., 2008) with a probe targeted to organisms in the candidate phylum OD1. The results revealed that the cells were very small (~0.3 μm) coccoid shape, and all of the stained cells were localized as aggregates on mineral particle-like structures (Figure 5a and Supplementary Figure 9). Scanning electron microscopy energy dispersive X-ray spectroscopy analysis of the CARD-FISH-stained samples showed that the substratum particles on which the cells were localized consisted primarily of silica, magnesium and iron (Figures 5b–h), consistent with them being in the group of serpentinite or peridotite minerals. These results suggested that the dominant organisms in the shallow and deep serpentinizing groundwater were associated with different minerals, both of which were abundantly present in the spring water.

Microscopic observation of OD1 organisms. (a) CARD-FISH detection of OD1 organisms (green fluorescence) that make aggregates in the mineral particles. (b–h) Scanning electron microscopy energy dispersive X-ray spectroscopy detection of the presence of Ca (b), Si (c), Fe (d), Mg (e), Cl (f), Na (g) and Cu (h) in the object that contains OD1 organisms. Blight (fluorescent) colors indicate the presence of minerals. The object contains silica (c), magnesium (d) and small amount of iron (f). Bars=5 μm.
Discussion
Metagenomics and genome-binning, taken together, have provided the genetic and genomic information that has helped to define the microbial communities associated with two ultrabasic springs (BS5 and GPS1), while revealing some highly unusual (perhaps unprecedented) features of the microbial community in the deep-water (GSP1) spring at The Cedars (Figure 1). Despite the similarity of the geochemistry, the metabolic capabilities of microbial communities associated with the two sources are very different. Hydrogen oxidation coupled with oxygen, nitrate or sulfate reduction is presumably a major energy metabolism in the community of shallow groundwater, while the energy strategies for the dominant deep community members remain elusive, with the possible exception of acetogenesis (for Chloroflexi) from molecular hydrogen and inorganic carbon (for example, bicarbonate or carbon monoxide). The dominant OCSs in the deep groundwater, in the candidate phylum OD1, have no respiratory genes, only a partial set of glycolysis genes, no ATP synthase genes (for four of the five OCSs) and no genes for the TCA cycle and the reductive acetyl-CoA pathway: how they generate ATP remains enigmatic. With regard to metabolism, they are similar to those of endosymbionts, but this seems unlikely since the metagenomic surveys failed to detect any evidence for eukaryotic organisms in the springs and the conditions of The Cedars’ springs should preclude any known eukaryote. The possibility remains that the OD1 organisms are intercellular symbionts of other members in the deep groundwater (Nelson and Stegen, 2015), such as the Chloroflexi, or that they are scavengers of dead cells, but further studies are needed to address this question. In any case, since the OD1 and Chloroflexi organisms consistently occupied over 90% of the deep community in the 2-year study (Figure 3b) and showed reasonable replication rate in this environment, both of those organisms should be actively growing in the deep groundwater environment. In addition, since closely related operational taxonomic units affiliated with the OD1 have been detected in the other serpentinization sites such as Lost City (Mid Atlantic Ridge) (Brazelton et al., 2010; Suzuki et al., 2013) and Prony Bay (New Caledonia) (Quéméneur et al., 2014), those members must be selected by the geological setting occurring at deep subsurface serpentinization globally.
The bin-genomes from GPS1 tend to have very small genomes (Figure 4). The reported genome sizes of other OD1 genomes are small in comparison with those of other Bacteria and Archaea (Wrighton et al., 2012; Kantor et al., 2013; Brown et al., 2015; Anantharaman et al., 2016). However, the OD1 genomes from GPS1 are even smaller: the smallest within the reported OD1 draft genomes that have been recovered mostly from acetate-amended neutral groundwater metagenomes (Wrighton et al., 2012; Brown et al., 2015; Anantharaman et al., 2016). Since other alkaliphiles have normal sized genomes (Takami et al., 2000; Muyzer et al., 2011; Zhao et al., 2011), it is unclear why the organisms at The Cedars maintain such reduced genome sizes. The combination of extremely low availability of energy (oxidant) and phosphorus may be the drivers that constrain genome sizes.
When the single-copy gene approach was used to assess the abundance of the various populations, an interesting difference was observed. The uncultured phylum OD1 was seen to be the dominant taxon, comprising over 70% of the community in the GPS1, and phylum Chloroflexi accounted for only ~20% of the community. This was in contrast to previous work, in which species abundance was estimated by 16S rRNA gene-based survey (Suzuki et al., 2013), and the phylum Chloroflexi was estimated to be the most abundant taxon (33–50%). The major reasons for the underestimation of the OD1 organisms via 16S rRNA gene-based survey are likely due to the presence of introns within the 16S rRNA gene (Supplementary Figure 4). Since the discovery of introns in the OD1 16S rRNA gene (Brown et al., 2015) and the potential risk of biases from PCR, the relative abundance of a microbial community is more accurately determined using single-copy housekeeping genes.
Since ATP synthases are generally thought to have evolved very early, the widespread distribution of diverse A-ATPases in bacterial phyla of The Cedars was unexpected. To our knowledge, the absence of F-ATPase genes in a bacteria-dominated microbial community has never been reported from any other environment. In fact, in the KEGG organisms (Moriya et al., 2007), 91% of bacterial genomes encode only F-ATPase (85%) or both F- and A-ATPases (6%) and bacterial genomes that encode only A-ATPase (7%) are limited to mainly three phyla (Chlamydia,Spirochaetes, Deinococcus-Thermus). On the other hand, with only two known exceptions, all archaeal genomes in the KEGG database encode only A-ATPases; Methanosarcina acetivorans and M. barkeri have both F- and A-ATPases. Notably, the previously sequenced genomes within both the phyla OD1 and Chloroflexi with the exceptions of two OD1 bin-genomes (Brown et al., 2015), and one Chloroflexi bin-genome RGB-2 (Hug et al., 2013), also encode only F-ATPase genes. These three exceptions were all retrieved from a metagenomic sequence of The Rifle Integrated Field Research, although most of the OD1 or Chloroflexi genomes from the Rifle site also encode F-ATPase genes. The phylogenetic analysis suggested that the A-ATPase genes in The Cedars deep community are most similar to those seen in archaeal or bacterial thermophiles in the phyla Crenarchaeota, Dictyoglomi or Caldiserica (Supplementary Figure 6), although none of these taxa were detected in the spring waters. Additionally, while members of the candidate phylum OD1 are known to have reduced metabolic potentials (Kantor et al., 2013; Nelson and Stegen, 2015), only two high-coverage genomes for OD1 have been identified to lack an entire ATPase gene set (Brown et al., 2015). In fact, with the exception of some insect endosymbionts that lack the entire gene set for ATP synthase (McCutcheon and Moran, 2012), all bacterial and archaeal isolates sequenced to date have full or partial sets of genes encoding F- and/or A-ATPase, except for the few OD1 organisms noted above. The well-studied intercellular symbiont Nanoarchaeum equitans (Waters et al., 2003), which is known for the absence of functional A-ATPase, also encodes part of the gene set for A-ATPase (ntpA, B, D, K) on the genome. It is very curious that four OD1 bin-genomes point to organisms having no detectable open reading frames indicating any known ATP synthase genes.
Based on the microbial studies of this site, the geochemical settings of the deep and shallow sources are expected to select for very different metabolic capabilities. Genomic evidence of the members in the shallow source indicates the capabilities of oxygen respiration, and the cultivated strains of Serpentinomonas, the dominant taxon in BS5, require low levels of both oxygen and calcium carbonate. Those facts may indicate that the shallow groundwater is a biosphere that interfaces the deep serpentinization and atmospheric environments on Earth. Meanwhile, the microbial community in the deep groundwater does not encode any respiration-related genes, and the dominant taxon OD1 is associated with particles of magnesium iron silicates (serpentine). Those results suggest that the deep community represents biomass being delivered from an active community deeper within the geological formation where the environment is highly reducing with no electron acceptors for respiration, and the community we see is metabolically linked to the active serpentinization community in the deep subsurface.
At this point, we can conclude that our genomic studies have revealed that while the harsh ultrabasic geological settings of The Cedars springs support multiple microbial populations, the deep-water-fed spring, GPS1, contains one of the most unusual communities ever reported. Analyses of metagenomes and bin-genomes reveal extremely small genomes, among the smallest ever reported. In addition, while this community is dominated by bacterial taxa, there is no evidence for the presence of bacterial (F-type) ATP synthase in the metagenome, and in those bacterial genomes that have recognizable ATP synthase genes, in every case they only have the genes encoding the A-type (archaeal) enzyme. The numerically dominant members of GPS1 are in the candidate phylum OD1, with very small genomes, often with no ATP synthase, with each OCS lacking one or more key biosynthetic pathways, suggesting that they are incapable of independent growth. The factors responsible for this unprecedented microbial community are not known. Clearly, it is not pH, as a nearby spring fed by shallow water (BS5) contains an alkaliphilic community dominated by Proteobacteria and has similar pH and Eh properties. The deep-water-fed spring, GPS1, may thus provide a special opportunity for understanding the evolution of genes, genomes, microorganisms and communities in deep subsurface environments, and perhaps early life on Earth.
References
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA . (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12: 402.
Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ et al. (2016). Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 7: 13219.
Barnes I, Lamarche Jr VC, Himmelberg G . (1967). Geochemical evidence of present-day serpentinization. Science 156: 830–832.
Brazelton WJ, Baross JA . (2009). Abundant transposases encoded by the metagenome of a hydrothermal chimney biofilm. ISME J 3: 1420–1424.
Brazelton WJ, Ludwig KA, Sogin ML, Andreishcheva EN, Kelley DS, Shen CC et al. (2010). Archaea and bacteria with surprising microdiversity show shifts in dominance over 1,000-year time scales in hydrothermal chimneys. Proc Natl Acad Sci USA 107: 1612–1617.
Brazelton WJ, Morrill PL, Szponar N, Schrenk MO . (2013). Bacterial communities associated with subsurface geochemical processes in continental serpentinite springs. Appl Environ Microbiol 79: 3906–3916.
Brazelton WJ, Nelson B, Schrenk MO . (2012). Metagenomic evidence for h(2) oxidation and h(2) production by serpentinite-hosted subsurface microbial communities. Front Microbiol 2: 268.
Brazelton WJ, Thornton CN, Hyer A, Twing KI, Longino AA, Lang SQ et al. (2016). Metagenomic identification of active methanogens and methanotrophs in serpentinite springs of the Voltri Massif, Italy. PeerJ Preprints 5: e2945.
Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A et al. (2015). Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523: 208–211.
Brown CT, Olm MR, Thomas BC, Banfield JF . (2016). Measurement of bacterial replication rates in microbial communities. Nat Biotechnol 34: 1256–1263.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85.
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Etiope G, Lollar BS . (2013). Abiotic methane on Earth. Rev Geophys 51: 276–299.
Hilario E, Gogarten JP . (1993). Horizontal transfer of ATPase genes—the tree of life becomes a net of life. Biosystems 31: 111–119.
Hoshino T, Yilmaz LS, Noguera DR, Daims H, Wagner M . (2008). Quantification of target molecules needed to detect microorganisms by fluorescence in situ hybridization (FISH) and catalyzed reporter deposition-FISH. Appl Environ Microbiol 74: 5068–5077.
Hug LA, Castelle CJ, Wrighton KC, Thomas BC, Sharon I, Frischkorn KR et al. (2013). Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22.
Ishii S, Suzuki S, Norden-Krichmar TM, Tenney A, Chain PSG, Scholz MB et al. (2013). A novel metatranscriptomic approach to identify gene expression dynamics during extracellular electron transfer. Nat Commun 4: 1601.
Ishii S, Suzuki S, Tenney A, Norden-Krichmar TM, Nealson KH, Bretschger O . (2015). Microbial metabolic networks in a complex electrogenic biofilm recovered from a stimulus-induced metatranscriptomics approach. Sci Rep 5: 14840.
Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ et al. (2013). Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. MBio 4: e00708–e00713.
Kawai M, Furuta Y, Yahara K, Tsuru T, Oshima K, Handa N et al. (2011). Evolution in an oncogenic bacterial species with extreme genome plasticity: Helicobacter pylori East Asian genomes. BMC Microbiol 11: 104.
Kohl L, Cumming E, Cox A, Rietze A, Morrissey L, Lang SQ et al. (2016). Exploring the metabolic potential of microbial communities in ultra-basic, reducing springs at The Cedars, CA, USA: experimental evidence of microbial methanogenesis and heterotrophic acetogenesis. J Geophys Res 121: 1203–1220.
Koonin EV, Martin W . (2005). On the origin of genomes and cells within inorganic compartments. Trends Genet 21: 647–654.
Li H, Durbin R . (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
McCutcheon JP, Moran NA . (2012). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26.
Miller HM, Matter JM, Kelemen P, Ellison ET, Conrad ME, Fierer N et al. (2016). Modern water/rock reactions in Oman hyperalkaline peridotite aquifers and implications for microbial habitability. Geochim Cosmochim Acts 179: 217–241.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M . (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–W185.
Morrill PL, Kuenen JG, Johnson OJ, Suzuki S, Rietze A, Sessions AL et al. (2013). Geochemistry and geobiology of a present-day serpentinization site in California: The Cedars. Geochim Cosmochim Acta 109: 222–240.
Mulkidjanian AY, Galperin MY, Koonin EV . (2009). Co-evolution of primordial membranes and membrane proteins. Trends Biochem Sci 34: 206–215.
Muller V, Gruber G . (2003). ATP synthases: structure, function and evolution of unique energy converters. Cell Mol Life Sci 60: 474–494.
Muyzer G, Sorokin DY, Mavromatis K, Lapidus A, Foster B, Sun H et al. (2011). Complete genome sequence of Thioalkalivibrio sp. K90mix. Stand Genomic Sci 5: 341–355.
Nelson WC, Stegen JC . (2015). The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol 6: 713.
Postec A, Quemeneur M, Bes M, Mei N, Benaissa F, Payri C et al. (2015). Microbial diversity in a submarine carbonate edifice from the serpentinizing hydrothermal system of the Prony Bay (New Caledonia) over a 6-year period. Front Microbiol 6: 857.
Quéméneur M, Bes M, Postec A, Mei N, Hamelin J, Monnin C et al. (2014). Spatial distribution of microbial communities in the shallow submarine alkaline hydrothermal field of the Prony Bay, New Caledonia. Environ Microbiol Rep 6: 665–674.
Quemeneur M, Palvadeau A, Postec A, Monnin C, Chavagnac V, Ollivier B et al. (2015). Endolithic microbial communities in carbonate precipitates from serpentinite-hosted hyperalkaline springs of the Voltri Massif (Ligurian Alps, Northern Italy). Environ Sci Pollut Res 22: 13613–13624.
Ragsdale SW, Pierce E . (2008). Acetogenesis and the Wood–Ljungdahl pathway of CO2 fixation. BBA Proteins Proteom 1784: 1873–1898.
Richter M, Rossello-Mora R . (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci USA 106: 19126–19131.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Sanchez-Murillo R, Gazel E, Schwarzenbach EM, Crespo-Medina M, Schrenk MO, Boll J et al. (2014). Geochemical evidence for active tropical serpentinization in the Santa Elena Ophiolite, Costa Rica: an analog of a humid early Earth? Geochem Geophys Geosyst 15: 1783–1800.
Schramm A, Fuchs BM, Nielsen JL, Tonolla M, Stahl DA . (2002). Fluorescence in situ hybridization of 16S rRNA gene clones (Clone-FISH) for probe validation and screening of clone libraries. Environ Microbiol 4: 713–720.
Schrenk MO, Brazelton WJ, Lang SQ . (2013). Serpentinization, carbon, and deep life. Carbon in Earth 75: 575–606.
Schulte M, Blake D, Hoehler T, McCollom T . (2006). Serpentinization and its implications for life on the early Earth and Mars. Astrobiology 6: 364–376.
Sleep NH, Bird DK, Pope EC . (2011). Serpentinite and the dawn of life. Philos Trans R Soc B 366: 2857–2869.
Sleep NH, Meibom A, Fridriksson T, Coleman RG, Bird DK . (2004). H2-rich fluids from serpentinization: geochemical and biotic implications. Proc Natl Acad Sci USA 101: 12818–12823.
Sorokin DY, Tourova TP, Mussmann M, Muyzer G . (2008). Dethiobacter alkaliphilus gen. nov sp nov., and Desulfurivibrio alkaliphilus gen. nov sp nov.: two novel representatives of reductive sulfur cycle from soda lakes. Extremophiles 12: 431–439.
Stamatakis A, Hoover P, Rougemont J . (2008). A rapid bootstrap algorithm for the RAxML Web servers. Syst Biol 57: 758–771.
Sugawara H, Ohyama A, Mori, Kurokawa K . (2009). Microbial Genome Annotation Pipeline (MiGAP) for diverse users. 20th International Conference on Genome Informatics. S-001: Kanagawa, Japan, pp 1–2.
Suzuki S, Ishii S, Wu A, Cheung A, Tenney A, Wanger G et al. (2013). Microbial diversity in The Cedars, an ultrabasic, ultrareducing, and low salinity serpentinizing ecosystem. Proc Natl Acad Sci USA 110: 15336–15341.
Suzuki S, Kuenen JG, Schipper K, van der Velde S, Ishii S, Wu A et al. (2014). Physiological and genomic features of highly alkaliphilic hydrogen-utilizing Betaproteobacteria from a continental serpentinizing site. Nat Commun 5: 3900.
Takami H, Nakasone K, Takaki Y, Maeno G, Sasaki R, Masui N et al. (2000). Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res 28: 4317–4331.
Takami H, Taniguchi T, Arai W, Takemoto K, Moriya Y, Goto S . (2016). An automated system for evaluation of the potential functionome: MAPLE version 2.1.0. DNA Res 23: 467–475.
Tiago I, Verissimo A . (2013). Microbial and functional diversity of a subterrestrial high pH groundwater associated to serpentinization. Environ Microbiol 15: 1687–1706.
von Ballmoos C, Cook GM, Dimroth P . (2008). Unique rotary ATP synthase and its biological diversity. Annu Rev Biophys 37: 43–64.
Waters E, Hohn MJ, Ahel I, Graham DE, Adams MD, Barnstead M et al. (2003). The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism. Proc Natl Acad Sci USA 100: 12984–12988.
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665.
Wu M, Eisen JA . (2008). A simple, fast, and accurate method of phylogenomic inference. Genome Biol 9: R151.
Zhao B, Mesbah NM, Dalin E, Goodwin L, Nolan M, Pitluck S et al. (2011). Complete genome sequence of the anaerobic, halophilic alkalithermophile Natranaerobius thermophilus JW/NM-WN-LF. J Bacteriol 193: 4023–4024.
Acknowledgements
We greatly appreciate Roger Raiche and David McCrory for providing their private land for our research. We thank Drs Greg Wanger, Orianna Bretschger and Nathan Walworth for the field support and constructive discussions. This work was funded by the NSF-EAR Grant Nos 1024872, 1424646 and 1424711. This work was partly supported by the JSPS Grand-in-Aid for Science Research Nos 26106004, 16K14647, 15H069907, 26251041 and 15K14907. This is a contribution to the Deep Carbon Observatory (DCO).
Author contributions
SS, SI and KHN designed research; SS, SI and AT performed metagenomic analyses, TH and FI performed microscopic analyses, AR and PLM performed aqueous geochemical analysis, SS, SI, JGK and KHN wrote the paper. SS and SI contributed to this research equally.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Suzuki, S., Ishii, S., Hoshino, T. et al. Unusual metabolic diversity of hyperalkaliphilic microbial communities associated with subterranean serpentinization at The Cedars. ISME J 11, 2584–2598 (2017). https://doi.org/10.1038/ismej.2017.111
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.111
This article is cited by
-
Comparative analysis of reconstructed ancestral proteins with their extant counterparts suggests primitive life had an alkaline habitat
Scientific Reports (2024)
-
Unique H2-utilizing lithotrophy in serpentinite-hosted systems
The ISME Journal (2023)
-
Diversification of methanogens into hyperalkaline serpentinizing environments through adaptations to minimize oxidant limitation
The ISME Journal (2021)
-
Microbial ecology of the newly discovered serpentinite-hosted Old City hydrothermal field (southwest Indian ridge)
The ISME Journal (2021)
-
Lipid analysis of CO2-rich subsurface aquifers suggests an autotrophy-based deep biosphere with lysolipids enriched in CPR bacteria
The ISME Journal (2020)
