
Abstract
Phylogeny is an ecologically meaningful way to classify plants and animals, as closely related taxa frequently have similar ecological characteristics, functional traits and effects on ecosystem processes. For bacteria, however, phylogeny has been argued to be an unreliable indicator of an organism’s ecology owing to evolutionary processes more common to microbes such as gene loss and lateral gene transfer, as well as convergent evolution. Here we use advanced stable isotope probing with 13C and 18O to show that evolutionary history has ecological significance for in situ bacterial activity. Phylogenetic organization in the activity of bacteria sets the stage for characterizing the functional attributes of bacterial taxonomic groups. Connecting identity with function in this way will allow scientists to begin building a mechanistic understanding of how bacterial community composition regulates critical ecosystem functions.
Similar content being viewed by others


Main
Microbiologists have hotly debated the relationship between phylogeny and function in microorganisms for decades. Significant genomic variation even within closely related organisms (that is, >97% 16S rRNA gene identity) suggests that even shallow clades may not be ecologically coherent (Doolittle and Zhaxybayeva, 2009). Further, bacterial strains with identical 16S rRNA gene sequences can exhibit distinct growth rates and substrate utilization profiles, suggesting limited niche overlap (Jaspers and Overmann, 2004). However, many functional genes are relatively conserved in prokaryotic phylogeny (Martiny et al., 2013). This conservation of gene content could underlie the accumulating evidence that phylogenetically clustered taxa are similar in their environmental distribution, niche space and functional capabilities, even at high levels of taxonomic organization (for example, order, class, phylum; Philippot et al., 2010; Lennon et al., 2012).
To date, evidence for phylogenetic organization of bacterial traits in intact assemblages has primarily relied on categorizing bacterial responses, such as changes in a taxon’s relative abundance (Placella et al., 2012; Evans and Wallenstein, 2014) or the presence in different environments (Morrissey and Franklin, 2015). Such research has provided early evidence that phylogeny can influence bacterial substrate utilization (Goldfarb et al., 2011; Mayali et al., 2014), habitat preference (Morrissey and Franklin, 2015) and life history strategies (Evans and Wallenstein, 2014). Although valuable, these assessments of bacterial traits are incomplete because they fail to capture quantitative variation among taxa. For instance, two taxa may be categorized as ‘cellulose utilizing’, but one may decompose and assimilate cellulose 10 times faster than the other, exerting more influence on cellulose decomposition rates. Rarely have quantitative measures of in situ activity been directly related to bacterial phylogeny (Amend et al., 2016); as a consequence, the significance of phylogeny in determining quantitative variation in microbial function is not well understood. This is likely because of the limited availability of techniques that can quantitatively measure taxon-specific activity. The current work uses a new technique, quantitative stable isotope probing (Hungate et al., 2015), to measure bacterial activity in soil. This technique works by determining the shift in the buoyant density of each taxon’s DNA caused by incorporation of a stable isotope tracer during incubation. Because the amount of isotope incorporation is directly proportional to the change in a given taxon’s buoyant density, this method enables quantitative measurement of isotope incorporation and thus bacterial activity.
This study aimed to test whether the activity of bacterial taxa in soil exhibits phylogenetic organization. We measured growth and glucose assimilation by exposing soil from a ponderosa pine forest in northern Arizona to 18O-labeled or unlabeled water for 7 days, with or without 13C or unlabeled glucose (n=3 per treatment, see Supplementary Materials for additional methodological details). Using quantitative stable isotope probing, the excess atom fraction 18O in the absence and presence of unlabeled glucose as well as the excess atom fraction 13C from glucose of each taxon’s DNA was quantified (Hungate et al., 2015). In brief, this technique uses density ultracentrifugation and high-throughput sequencing to measure changes in the density of microbial DNA following exposure to a heavy isotope. By modeling the effects of genomic GC content and isotope incorporation on the DNA molecular weight and density, this technique can quantitatively estimate isotope incorporation into DNA irrespective of nucleic acid composition. Because water is a universal substrate for DNA synthesis (Schwartz, 2007), a taxon’s genomic 18O content reflects its population growth during the incubation period. Similarly, genomic 13C content provides a quantitative measure of each taxon’s carbon assimilation from glucose. To determine whether these activities exhibited phylogenetic organization, we tested for a phylogenetic signal, which is the tendency of related organisms to resemble each other more than would be expected by chance (Pagel, 1999; Blomberg et al., 2003).
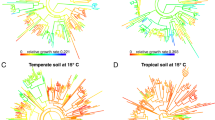
All three measures of bacterial activity, growth with and without added glucose as well as carbon assimilation from glucose, were clustered across the 16S rRNA gene-based phylogenetic tree (Figure 1,Table 1). These results provide robust evidence that bacterial evolutionary history has a measurable influence on the activity of bacteria in intact soil communities. Our estimates of λ approached 1, signifying that the variation in bacterial activity was approximately proportional to what would be expected under a neutral drift model of evolution (Freckleton et al., 2002). However, the more conservative Blomberg’s K statistics were <1, suggesting that relatives were less similar than would be expected under neutral drift (Blomberg et al., 2003). Overall, we detected phylogenetic signals that were similar in strength to those reported for a variety of ecological traits in plants and animals, such as habitat breadth and fecundity (Freckleton et al., 2002; Blomberg et al., 2003).

Phylogenetic tree (based on 16S rRNA gene sequences) and isotope incorporation of bacterial taxa in soil. Bars are proportional to the excess atom fraction of 18O or 13C of each taxon’s DNA after incubation with 13C-glucose (blue) or H218O in the presence (green) or absence (red) of natural abundance glucose. Tree is colored by the phylogenetic group.
Examining within- versus among-group variation across bacterial taxonomy also shows evidence of this phylogenetic organization. Taxonomic group, from family to phylum, explained a significant amount of the variation in 18O or 13C excess atom fraction across all taxonomic levels (Figure 2, P<0.001 in all cases). The finding that microbial activities are clustered at the phylum level suggests that variation in bacterial activities arise from physiological and ecological differences deeply rooted in bacterial phylogeny. As would be expected, group identity at finer levels of taxonomic resolution is able to explain increasing amounts of variation in bacterial activity. Family membership explained the majority of variation in growth (that is, 18O incorporation) and carbon incorporation, reflecting the significant differences in the activities of bacterial families (Supplementary Figure S1). For example, all members of the Micrococcaceae family exhibited high 13C assimilation from glucose (ranging from 0.41 to 0.52 13C excess atom fraction), while assimilation by members of the Solibacteraceae was much lower (ranging from 0.04 to 0.06 13C excess atom fraction). These findings are consistent with patterns documented in plants, where phylogenetic groups exhibit distinct functional traits, such as leaf size and seed mass (Cornwell et al., 2008).

Variation in excess atom fraction of 18O or 13C of each taxon’s DNA after incubation with 13C-glucose (blue) or H218O in the presence (green) or absence (red) of natural abundance glucose explained by group membership at different taxonomic levels. Values reflect linear model results for groups with a minimum of five member taxa.
The ability to use simple carbon substrates is common in bacteria and has been suggested to display only shallow phylogenetic clustering (Martiny et al., 2013), an indication that microbial taxa are functionally redundant with regard to the processing of these compounds (McGuire and Treseder, 2010; Martiny et al., 2013). If the hypothesis of functional redundancy were true, one would expect little-to-no difference in carbon metabolism or assimilation across the bacterial phylogeny, which would produce a weak phylogenetic signal. Our results challenge this notion of functional redundancy: although nearly all organisms incorporated 13C, the degree of enrichment differed dramatically across taxa and families. Thus, even though the ability to assimilate simple carbon substrates in pure culture may be relatively ubiquitous and display only shallow clustering, the rates of carbon assimilation in situ may vary significantly across bacterial phylogeny. In an intact community, carbon assimilation rates likely depend on a variety of ecological traits and physiological systems, such as those governing the transport of substrates into the cell and the organism’s growth rate. Our findings highlight potential pitfalls with using qualitative measures, such as genetic potential or substrate utilization tests (Martiny et al., 2013), to infer functional redundancy, which is defined as a quantitative equivalency in the functioning of taxa (Allison and Martiny, 2008).
The variable growth (that is, 18O assimilation) observed here could reflect differing trophic strategies across bacterial taxa. Growth rate, especially in a nutrient-rich environment, is a defining characteristic of trophic strategy, where relatively rapid growth suggests copiotrophy and slow growth indicates oligotrophy (Lauro et al., 2009). Trophic strategy has been linked to many genomic features in bacteria, such as genome size, rRNA operon number and abundance of genes encoding for periplasmic membrane proteins (Lauro et al., 2009). It is likely that a combination of such genomic features underlie the variation in growth observed here. Growth is a process that involves, and is regulated by, many gene systems in bacteria, resulting in phenotypes that reflect an integration of many individual processes. This complexity could be one reason why the phylogenetic signal was so robust, as many gains, losses and changes to individual genes would be required to transition a slow-growing bacterium into a fast-growing bacterium.
Evaluating phylogenetic organization, as carried out in the current work, will allow more accurate characterization of phylogenetic groups. For instance, prior studies have described Actinobacteria as both oligotrophic (Bastian et al., 2009) and copiotrophic (Leff et al., 2015). Such conflicting results highlight the imprudence of describing high-level taxonomic groups without assessing whether the responses of member taxa are coherent. Our results indicate that Actinobacteria contains both copiotrophic and oligotrophic families (for example, Micrococcaceae and Rubrobacteraceae, respectively). Thus Actinobacteria could be dominated by copiotrophic taxa in one environment and oligotrophic taxa in another. As a consequence, generalizations about trophic strategy at the phylum level may not be possible or appropriate. Although our results are in agreement with some proposed classifications in the literature, such as β- and γ-Proteobacteria as being relatively copiotrophic (Eilers et al., 2010), we suggest researchers evaluate and describe the phylogenetic organization associated with an ecological trait or activity before making generalizations to an entire phylogenetic group.
Microbial community composition affects ecosystem functioning from litter decomposition (Allison et al., 2013) to plant fitness (Lau and Lennon, 2012). However, the mechanisms of these effects are unknown because very little information is available on the distribution of in situ activities across prokaryotes. This lack of information has prevented microbial ecologists from testing important questions about the functional redundancy of microorganisms in their natural communities. If bacterial taxa perform a process at the same rate under the same environmental conditions, thereby making them functionally redundant, then a change in the diversity of that community is unlikely to affect the community-level process rate (Allison and Martiny, 2008). However, we found individual taxa and phylogenetic groups to be functionally dissimilar with respect to growth and carbon assimilation, providing a basis for functional differences among phylogenetically distinct communities. Consequently, this research sets the stage for characterizing the functions of microbial taxa and phylogenetic groups in order to build a mechanistic understanding of how phylogenetic community composition influences ecosystem processes.
References
Allison SD, Lu Y, Weihe C, Goulden ML, Martiny AC, Treseder KK et al. (2013). Microbial abundance and composition influence litter decomposition response to environmental change. Ecology 94: 714–725.
Allison SD, Martiny JB . (2008). Resistance, resilience, and redundancy in microbial communities. Proc Natl Acad Sci USA 105: 11512–11519.
Amend AS, Martiny AC, Allison SD, Berlemont R, Goulden ML, Lu Y et al. (2016). Microbial response to simulated global change is phylogenetically conserved and linked with functional potential. ISME J 10: 109–118.
Bastian F, Bouziri L, Nicolardot B, Ranjard L . (2009). Impact of wheat straw decomposition on successional patterns of soil microbial community structure. Soil Biol Biochem 41: 262–275.
Blomberg SP, Garland Jr T, Ives AR . (2003). Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution 57: 717–745.
Cornwell WK, Cornelissen JHC, Amatangelo K, Dorrepaal E, Eviner VT, Godoy O et al. (2008). Plant species traits are the predominant control on litter decomposition rates within biomes worldwide. Ecol Lett 11: 1065–1071.
Doolittle WF, Zhaxybayeva O . (2009). On the origin of prokaryotic species. Genome Res 19: 744–756.
Eilers KG, Lauber CL, Knight R, Fierer N . (2010). Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Biol Biochem 42: 896–903.
Evans SE, Wallenstein MD . (2014). Climate change alters ecological strategies of soil bacteria. Ecol Lett 17: 155–164.
Freckleton RP, Harvey PH, Pagel M . (2002). Phylogenetic analysis and comparative data: a test and review of evidence. Am Nat 160: 712–726.
Goldfarb KC, Karaoz U, Hanson CA, Santee CA, Bradford MA, Treseder KK et al. (2011). Differential growth responses of soil bacterial taxa to carbon substrates of varying chemical recalcitrance. Front Microbiol 2: 94.
Hungate BA, Mau RL, Schwartz E, Caporaso JG, Dijkstra P, van Gestel N et al. (2015). Quantitative microbial ecology through stable isotope probing. Appl Environ Microbiol 81: 7570–7581.
Jaspers E, Overmann J . (2004). Ecological significance of microdiversity: identical 16S rRNA gene sequences can be found in bacteria with highly divergent genomes and ecophysiologies. Appl Environ Microbiol 70: 4831–4839.
Lau JA, Lennon JT . (2012). Rapid responses of soil microorganisms improve plant fitness in novel environments. Proc Natl Acad Sci USA 109: 14058–14062.
Lauro FM, McDougald D, Thomas T, Williams TJ, Egan S, Rice S et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc Natl Acad Sci USA 106: 15527–15533.
Leff JW, Jones SE, Prober SM, Barberán A, Borer ET, Firn JL et al. (2015). Consistent responses of soil microbial communities to elevated nutrient inputs in grasslands across the globe. Proc Natl Acad Sci USA 112: 10967–10972.
Lennon JT, Aanderud ZT, Lehmkuhl BK, Schoolmaster DR Jr . (2012). Mapping the niche space of soil microorganisms using taxonomy and traits. Ecology 93: 1867–1879.
Martiny AC, Treseder K, Pusch G . (2013). Phylogenetic conservatism of functional traits in microorganisms. ISME J 7: 830–838.
Mayali X, Weber PK, Mabery S, Pett-Ridge J . (2014). Phylogenetic patterns in the microbial response to resource availability: amino acid incorporation in San Francisco bay. PLoS One 9: e95842.
McGuire KL, Treseder KK . (2010). Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol Biochem 42: 529–535.
Morrissey EM, Franklin RB . (2015). Evolutionary history influences the salinity preference of bacterial taxa in wetland soils. Front Microbiol 6: 1013.
Pagel M . (1999). Inferring the historical patterns of biological evolution. Nature 401: 877–884.
Philippot L, Andersson SG, Battin TJ, Prosser JI, Schimel JP et al. (2010). The ecological coherence of high bacterial taxonomic ranks. Nat Rev Microbiol 8: 523–529.
Placella SA, Brodie EL, Firestone MK . (2012). Rainfall-induced carbon dioxide pulses result from sequential resuscitation of phylogenetically clustered microbial groups. Proc Nat Acad Sci 109: 10931–10936.
Schwartz E . (2007). Characterization of growing microorganisms in soil by stable isotope probing with H218O. Appl Environ Microbiol 73: 2541–2546.
Acknowledgements
This research was supported by grants from the National Science Foundation (EAR-1124078 and DEB-1321792) and the Department of Energy's Biological Systems Science Division, Program in Genomic Science.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Morrissey, E., Mau, R., Schwartz, E. et al. Phylogenetic organization of bacterial activity. ISME J 10, 2336–2340 (2016). https://doi.org/10.1038/ismej.2016.28
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.28
This article is cited by
-
The predictive power of phylogeny on growth rates in soil bacterial communities
ISME Communications (2023)
-
Life history strategies among soil bacteria—dichotomy for few, continuum for many
The ISME Journal (2023)
-
Nutrients strengthen density dependence of per-capita growth and mortality rates in the soil bacterial community
Oecologia (2023)
-
Enhanced carbon, nitrogen and associated bacterial community compositional complexity, stability, evenness, and differences within the tree-soils of Inga punctata along an age gradient of planted trees in reforestation plots
Plant and Soil (2023)
-
Unbalanced diets enhance the complexity of gut microbial network but destabilize its stability and resistance
Stress Biology (2023)
