
Abstract
The cellulolytic protist Trichonympha agilis in the termite gut permanently hosts two symbiotic bacteria, ‘Candidatus Endomicrobium trichonymphae’ and ‘Candidatus Desulfovibrio trichonymphae’. The former is an intracellular symbiont, and the latter is almost intracellular but still connected to the outside via a small pore. The complete genome of ‘Ca. Endomicrobium trichonymphae’ has previously been reported, and we here present the complete genome of ‘Ca. Desulfovibrio trichonymphae’. The genome is small (1 410 056 bp), has many pseudogenes, and retains biosynthetic pathways for various amino acids and cofactors, which are partially complementary to those of ‘Ca. Endomicrobium trichonymphae’. An amino acid permease gene has apparently been transferred between the ancestors of these two symbionts; a lateral gene transfer has affected their metabolic capacity. Notably, ‘Ca. Desulfovibrio trichonymphae’ retains the complex system to oxidize hydrogen by sulfate and/or fumarate, while genes for utilizing other substrates common in desulfovibrios are pseudogenized or missing. Thus, ‘Ca. Desulfovibrio trichonymphae’ is specialized to consume hydrogen that may otherwise inhibit fermentation processes in both T. agilis and ‘Ca. Endomicrobium trichonymphae’. The small pore may be necessary to take up sulfate. This study depicts a genome-based model of a multipartite symbiotic system within a cellulolytic protist cell in the termite gut.
Similar content being viewed by others

Introduction
Termites require symbioses with gut microbes, in order to digest dead plant matter and obtain nitrogenous compounds (Brune, 2014; Hongoh, 2011). Members of phylogenetically basal (‘lower’) termite taxa harbour in their guts a dense community of protists, bacteria and archaea. The protists generally establish a symbiotic relationship with multiple species of prokaryotes, which reside in their cytoplasm, nucleoplasm or attach onto the cell surface (Brune, 2014; Sato et al., 2014). Although metagenome, metatranscriptome and metabolome analyses of the microbiota in the gut of lower termites have been performed (Tartar et al., 2009; Do et al., 2014; Tokuda et al., 2014), the functions of individual microbial species and their interrelationships mostly remain unclear. In particular, the multipartite symbiotic system comprising cellulolytic protists and their multiple prokaryotic endo- and/or ectosymbionts has not been characterized in detail.
Trichonympha agilis, a cellulolytic parabasalid protist that is present in the gut of the termite Reticulitermes speratus, hosts two bacterial symbionts, ‘Candidatus Endomicrobium trichonymphae’ phylotype Rs-D17 (class Endomicrobia; Stingl et al., 2005; Ohkuma et al., 2007) and ‘Candidatus Desulfovibrio trichonymphae’ phylotype Rs-N31 (class Deltaproteobacteria; Sato et al., 2009). The cellular association between T. agilis and these two bacteria species is permanent: ca. 4,000 and 1,800 cells of ‘Ca. Endomicrobium trichonymphae’ and ‘Ca. Desulfovibrio trichonymphae’, respectively, always inhabit the T. agilis cell in specific subcellular locations, as shown in Figures 1a and b (Sato et al., 2009). These bacteria account for ca. 4% and 2% of the total prokaryotic cells in the R. speratus gut, respectively (Sato et al., 2009).

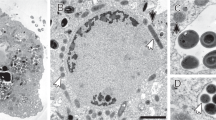
Fluorescence in situ hybridization (FISH) and transmission electron microscopy (TEM) of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 cells associated with a Trichonympha agilis cell. (a) Phase-contrast image of T. agilis. (b) FISH analysis using oligonucleotide probes specific to ‘Ca. Desulfovibrio trichonymphae’ (6-carboxyfluorescein-labelled, green) and to ‘Ca. Endomicrobium trichonymphae’ (Texas red-labelled, red), respectively (Sato et al., 2009). (c) TEM image of ‘Ca. Desulfovibrio trichonymphae’ cells in a T. agilis cell. Pores opening to the outside of the T. agilis cell were observed (arrows). H, hydrogenosome. (d) Magnified image of a ‘Ca. Desulfovibrio trichonymphae’ cell in panel c. IM, inner membrane; OM, outer membrane; HPM, host plasma membrane. Bars: 20 μm (a, b); 500 nm (c); 100 nm (d).
The complete genome sequence of the uncultured, intracellular symbiont ‘Ca. Endomicrobium trichonymphae’ (1.15 Mb, including plasmids) was previously obtained using a whole genome amplification (WGA) technique (Hongoh et al., 2008a). Its small genome showed the potential to synthesize various amino acids and cofactors and to ferment monosaccharides to acetate, lactate, ethanol, CO2 and H2 (Hongoh et al., 2008a).
‘Ca. Desulfovibrio trichonymphae’ is uncultured and was previously considered to be an intracellular symbiont (Sato et al., 2009). However, transmission electron microscopy (TEM) of Trichonympha globulosa in the gut of the termite Incisitermes marginipennis revealed that its Desulfovibrio symbionts are localized in deep invaginations of the host (Trichonympha) plasma membrane that are open to the exterior of the host cell (Strassert et al., 2012). Our re-examination showed that ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 cells were almost completely buried in the host cytoplasm but still connected to the outside through a small pore (Figures 1c and d). Analyses using the reverse transcription polymerase chain reaction (RT-PCR) showed that ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 transcribed the dsrAB and apsA genes that are responsible for sulfate reduction and hynA for hydrogen oxidation (Sato et al., 2009). No other information on the functions of ‘Ca. Desulfovibrio trichonymphae’ has been available hitherto.
In this study, we attempted to acquire the complete genome sequence of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31, in order to predict its functions and roles in the symbioses with T. agilis and ‘Ca. Endomicrobium trichonymphae’ phylotype Rs-D17. Our results provide a genome-based model of a tripartite symbiotic system within a cellulolytic protist cell in the termite gut.
Materials and methods
Termites, Fluorescence in situ hybridization (FISH) and TEM analysis
The wood-feeding termite R. speratus (family Rhinotermitidae) was collected in Saitama Prefecture, Japan. After rearing with cellulose powder for three days, worker termites were subjected to experiments. FISH and TEM were performed as described previously (Sato et al., 2009, 2014). The wood-feeding termite Hodotermopsis sjostedti (family Termopsidae) was collected in Kagoshima Prefecture, Japan.
Collection of ‘Ca. Desulfovibrio trichonymphae’ cells and WGA
The gut of R. speratus was removed from a worker termite, and the gut contents were suspended in buffer solution U (Trager, 1934). A single cell of T. agilis was physically isolated using a TransferMan NK2 micromanipulator (Eppendorf, Hamburg, Germany) with a glass capillary. The collected T. agilis cell was washed several times in buffer and dissected into the anterior and posterior parts using the micromanipulator equipped with a Feather blade handle MF-130S and a K-730 micro-blade. The anterior part containing ‘Ca. Desulfovibrio trichonymphae’ cells was collected in a 0.2 ml PCR tube and subjected to WGA, using the illustra GenomiPhi HY DNA Amplification Kit (GE Healthcare, Little Charlfont, UK), as described previously (Hongoh et al., 2008a, 2008b).
Genome analysis
Sequencing libraries for the ‘Ca. Desulfovibrio trichonymphae’ genome were prepared using the TruSeq DNA PCR-free Sample Prep Kit and the Nextera Mate Pair Sample Prep Kit (Illumina, San Diego, CA, USA). Sequencing was performed using the MiSeq Reagent Kit v3 (600 cycles) on an Illumina MiSeq platform. The generated reads were processed for adapter and quality trimming using programs cutadapt and prinseq, respectively (Martin, 2011; Schmieder and Edwards, 2011). The reads were assembled into contigs, using SPAdes 3.0 (Bankevich et al., 2012), and the contigs and the mate-pair reads were used to generate scaffolds with SCARPA 0.241 (Donmez and Brudno, 2013). Finding and functional annotation of genes were performed using MiGAP (http://www.migap.org), and the result was curated manually. Pseudogenes were manually identified as described previously (Hongoh et al., 2008a). Metabolic pathways were reconstructed using the KEGG automatic annotation server (KAAS; Moriya et al., 2007). Genes were assigned to functional categories based on non-supervised orthologous groups (NOG) (Powell et al., 2014). Clustered regularly interspaced short palindromic repeat (CRISPR) loci were identified using CRISPRFinder (Grissa et al., 2007).
RT-PCR
Gut contents of ten R. speratus workers were suspended in solution U in a 1.5 ml tube, and centrifuged at a low speed to collect cells of large protists including T. agilis. The sedimented cells were subjected to RNA extraction, using the RNA PowerViral Environmental RNA/DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, USA), and contaminating DNA was digested using the TURBO DNA-free Kit (Ambion, Waltham, MA, USA). Extracted RNA was reverse-transcribed using the SuperScript III Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA). Generated cDNA was used as template for PCR amplification, using the Phusion High-Fidelity DNA Polymerase (NEB) with primers listed in Supplementary Table S1. No specific amplification was detected from the RNA samples without reverse transcription in RT-PCR. Amplification products were cloned using the Zero Blunt TOPO PCR Cloning Kit for Sequencing (Invitrogen) and Competent Quick DH5α (TOYOBO, Tokyo, Japan), and were sequenced using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosciences, Foster City, CA, USA) on an ABI 3730xl DNA Analyzer, as described previously (Sato et al., 2014).
Phylogenetic analysis and codon adaptation index (CAI) calculation
Maximum-likelihood trees were constructed using MEGA6 (Tamura et al., 2013). Sequences were aligned using MUSCLE (Edgar, 2004) with manual corrections, and ambiguously aligned sites were removed using Gblocks (Talavera and Castresana, 2007). CAI was calculated using CAIcal (Puigbo et al., 2008). The gene coding for AroP was identified by a BLAST search using the aroP gene of ‘Ca. Endomicrobium trichonymphae’ phylotype Rs-D17 as the query, in our ongoing genome analysis of ‘Candidatus Endomicrobium sp.’ HsTcC-Em16 from a single cell of Trichonympha sp. HsjTcC in the gut of the termite H. sjostedti.
Accession numbers
The sequences reported in this study have been deposited in DDBJ under accession numbers AP017368 for the ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 genome and LC122271 for aroP of ‘Ca. Endomicrobium sp.’ HsTcC-Em16.
Results
Morphology of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31
We re-investigated the morphology of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 associated with T. agilis cells in the gut of R. speratus. TEM analysis showed that the majority of the ‘Ca. Desulfovibrio trichonymphae’ cells were coccoid, embedded in the peripheral region of the host cytoplasm, and connected to the outside through a small pore with 41.4±5.1 nm diameter (mean±s.d., n=14; Figures 1c and d). When ‘Ca. Desulfovibrio trichonymphae’ cells were present deeper inside the host cytoplasm, the surrounding host membrane extended like tubes, which connected adjacent ‘Ca. Desulfovibrio trichonymphae’ cells to each other (Supplementary Figure S1). The tube-like structures possibly lead to the exterior, although not confirmed. The bacterium possessed inner and outer membranes, and its lipopolysaccharide layer was not prominent (Figure 1d).
General features of the ‘Ca. Desulfovibrio trichonymphae’ genome
We reconstructed the complete genome sequence of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 with no gaps or ambiguous nucleotide sites, from a WGA product of the anterior part of a single T. agilis cell. The genome consisted of a circular chromosome of 1 410 056 bp (Supplementary Figure S2); no plasmids were found. The chromosome contains 1082 putative protein-coding sequences (CDSs), two rRNA operons, and 49 tRNA genes corresponding to codons for all 20 amino acids (Table 1). In addition, 188 pseudogenes were identified (Supplementary Table S2) and classified into NOG (Supplementary Figure S3). Of the 188 pseudogenes, 55 (29.3%) were assigned to category [L] (replication, recombination and repair) including transposase, integrase, DNA methyltranferase and CRISPR-associated proteins, and 14 (7.4%) were to category [V] (defense mechanisms) including DNA restriction-modification systems (Supplementary Figure S3). A remnant of the CRISPR region was identified. The biosynthetic pathway for lipidA is missing as seen in a free-living relative, Desulfovibrio desulfuricans ATCC 27774, and several other genes involved in lipopolysaccharide biosynthesis are pseudogenized (Supplementary Table S2).
A maximum-likelihood tree based on concatenated sequences of 30 ribosomal proteins showed that D. desulfuricans ATCC 27774 was the closest relative to ‘Ca. Desulfovibrio trichonymphae’ among the genome-sequenced bacteria, and these two bacteria clustered with Lawsonia intracellularis PHE/MN1-00, which is an intracellular pathogen of swine intestine that causes proliferative enteropathy (Mcorist et al., 1995; Supplementary Figure S4). The genome size of ‘Ca. Desulfovibrio trichonymphae’ is approximately half that of D. desulfuricans ATCC 27774, and similar to that of L. intracellularis PHE/MN1-00 (Table 1). The G+C content (55%) was comparable with that of D. desulfuricans ATCC 27774 and much higher than that of L. intracellularis PHE/MN1-00 (Table 1).
Carbon and energy metabolism
Predicted metabolic pathways of ‘Ca. Desulfovibrio trichonymphae’ are outlined in Figure 2. The genome retains pathways for gluconeogenesis, non-oxidative pentose phosphate biosynthesis, and a partial tricarboxilic acid (TCA) cycle, allowing the biosynthesis of various compounds including amino acids, cofactors, nucleotides, and peptidoglycans. In addition, ‘Ca. Desulfovibrio trichonymphae’ encodes genes for tricarboxylate transporter (TctABC), which possibly imports citrate (Winnen et al., 2003). Citrate can be converted to 2-oxoglutarate, which is a precursor of amino acids.

Predicted metabolic pathways of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31. Genes responsible for the pathways marked with red X’s are pseudogenized.
Genes coding for glucokinase, diphosphate-fructose-6-phosphate 1-phosphotransferase, and pyruvate kinase are pseudogenized; the bacterium has lost the glycolytic pathway (Figure 2). Consistently, it lacks genes for mannose permease, which are commonly found in the genomes of desulfovibrios (Santana and Crasnier-Mednansky, 2006). In addition, the genome lacks the genes for lactate dehydrogenases, and genes for lactate utilization proteins (LutABC) and lactate permease are pseudogenized; thus, it cannot use lactate as an electron donor nor a carbon source although this ability is common in desulfovibrios (Heidelberg et al., 2004; Keller and Wall, 2011). A gene coding for a candidate pyruvate transporter, LctP-2 (Meyer et al., 2014), is absent. Pathways for using malate, fumarate, and/or succinate as carbon sources were not found. Since the genome encodes genes involved in conversion of acetate to acetyl-CoA, and acetyl-CoA plus CO2 to pyruvate, the main carbon sources appear to be acetate and CO2 (Badziong et al., 1979). Genes coding for the complete components of FoF1-ATPase, dissimilatory sulfite reductase, fumarate reductase and several hydrogenases were found, while genes for formate dehydrogenase and alcohol dehydrogenase are absent. These indicate that the bacterium generates energy via anaerobic respiration using H2 as the electron donor and sulfate and/or fumarate as the electron acceptors (Figures 2 and 3).

Predicted electron flows of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31. Hmc, high-molecular-weight cytochrome c; c3, cytochrome c class III; MQ, menaquinone; Dsr, dissimilatory sulfite reductase; Qmo, quinone-modifying oxidoreductase; Apr, adenylylsulfate reductase; APS, adenylyl sulfate; Hdr, heterodisulfide reductase; Flx, NAD(P)H dehydrogenase; Nfn, ferredoxin-NADP(+) reductase; Coo-Hase, CO-induced membrane-bound hydrogenase; Ech-Hase, energy-conserving membrane-bound hydrogenase; Nrf, cytochrome c nitrite reductase; Frd, fumarate reductase; DcuA, anaerobic C4-dicarboxylate antiporter.
Hydrogen metabolism
Predicted mechanisms to oxidize H2 in ‘Ca. Desulfovibrio trichonymphae’ are shown in Figure 3. ‘Ca. Desulfovibrio trichonymphae’ has genes for three hydrogenases: periplasmic (NiFe) hydrogenase (HynAB) and two membrane-bound H+-translocating (NiFe) hydrogenase complexes (CooFHKLMUX and EchABCDEF) that have active sites facing the cytoplasm. Considering the consistent supply of H2 from the host hydrogenosomes and formation of H+-membrane potential by the action of HynAB, it is likely that one or both of the membrane-bound hydrogenases oxidize H2 with inward H+-translocation (Pereira et al., 2011). This should be coupled with the formation of a reduced form of ferredoxin and/or nicotinamide adenine dinucleotide (NADH), which are required to fix CO2 and also possibly to reduce sulfate (Figure 3; Ramos et al., 2015). Genes for (FeFe) hydrogenase and (NiFeSe) hydrogenase are absent (Supplementary Tables S2 and S3).
The genome encodes genes for periplasmic cytochrome c class III (c3), membrane-bound high-molecular-weight cytochrome c (HmcABCDEF) and membrane-bound Hdr-like menaquinol oxidoreductase (DsrMKJOP). These transmit electrons produced by the action of HynAB to the dissimilatory sulfite reductase subunit DsrC (Figure 3; Keller and Wall, 2011). The genome possesses genes for the biosynthesis of menaquinone, which is an electron carrier in the plasma membrane.
‘Ca. Desulfovibrio trichonymphae’ retains genes for sodium:sulfate symporter, dissimilatory sulfite reductase (DsrAB), adenylylsulfate reductase (AprAB), quinone-modifying oxidoreductase (QmoABC), heterodisulfide reductase (HdrABC) and Hdr-coupled NADH dehydrogenase (FlxABCD) (Ramos et al., 2015) (Figure 3 and Supplementary Table S3). Thus, ‘Ca. Desulfovibrio trichonymphae’ most likely has the ability to reduce sulfate to sulfide, which can be assimilated into cysteine and/or diffused toward the outside of the cell (Grein et al., 2013). In addition, the bacterium has genes for membrane-bound cytochrome c nitrite reductase (NrfAH), which protects sulfate-reducing activity from strong inhibition caused by nitrite (Greene et al., 2003). The genome possesses genes for fumarate reductase (FrdABCD), fumarase, aspartase, and anaerobic C4-dicarboxylate (fumarate, succinate, malate and aspartate) antiporter (DcuA). Since in other parabasalids, such as Trichomonas vaginalis, malate is produced in the cytoplasm as a metabolic intermediate in its fermentation process (Müller et al., 2012), ‘Ca. Desulfovibrio trichonymphae’ possibly takes up malate from the host T. agilis cell, converts it to fumarate, conducts fumarate respiration, and exports succinate (Figure 3; Ullmann et al., 2000).
We performed RT-PCR for cooH, echE, hdrA, frdA and nrfA, to verify expressions of each of these genes. All of their transcripts were detected. Transcripts of the dsrAB, aprA (apsA) and hynA genes were detected previously (Sato et al., 2009). Thus, the sulfate-reducing system using H2 as an electron donor is probably functional. The gene for the redox-sensing transcriptional repressor Rex, which downregulates the expression of genes and operons for sulfate respiration (Ravcheev et al., 2012; Christensen et al., 2015), is pseudogenized. The predicted regulon governed by Rex in desulfovibrios includes genes for sulfate adenyltransferase and adenylate kinase, and operons for dsrAB, aprAB, dsrMKJOP, qmoABC, cooFHKLMUX and FoF1-ATP synthase subunits, according to the RegPrecise database (Novichkov et al., 2013). These genes might be constitutively expressed.
Biosynthesis of amino acids, cofactors and nucleotides
‘Ca. Desulfovibrio trichonymphae’ retains biosynthetic pathways for 18 amino acids and possesses genes for transporters of methionine (MetNIQ), aromatic amino acids (AroP), and branched-chain amino acids and possibly threonine (LivKHMGF) (Figure 2). Among the 18 amino acids, five (asparagine, cystein, glutamine, proline and serine) cannot be synthesized by ‘Ca. Endomicrobium trichonymphae’ (Hongoh et al., 2008a). Conversely, ‘Ca. Desulfovibrio trichonymphae’ does not possess biosynthetic pathways for methionine and threonine, both of which can be provided by ‘Ca. Endomicrobium trichonymphae’ (Hongoh et al., 2008a); thus, the capacity of amino acid biosynthesis is partially complementary (Supplementary Table S4). Furthermore, ‘Ca. Desulfovibrio trichonymphae’ has the potential to synthesize various cofactors, including heme and cobalamin (vitamin B12) (Figure 2). The cofactor-biosynthesis capacity is also partially complementary with that of ‘Ca. Endomicrobium trichonymphae’ (Supplementary Table S5). ‘Ca. Desulfovibrio trichonymphae’ retains nucleotide biosynthetic pathways. Genes for nitrogen fixation and transporters for ammonium or urea are absent.
Comparative genome analysis
We classified genes into NOG and compared the genome of ‘Ca. Desulfovibrio trichonymphae’ with D. desulfuricans ATCC 27774, L. intracellularis PHE/MN1-00, and ‘Ca. Endomicrobium trichonymphae’ (Figure 4 and Supplementary Figure S5). As in ‘Ca. Endomicrobium trichonymphae’, the number of genes involved in signal transduction is reduced, and genes for motility and chemotaxis are missing or pseudogenized; ‘Ca. Desulfovibrio trichonymphae’ has lost its motility, which is normally a characteristic of the genus Desulfovibrio including L. intracellularis. ‘Ca. Desulfovibrio trichonymphae’ should thus be vertically transmitted and have no free-living phase. The proportion of genes categorized in energy production and conversion (C), amino acid transport and metabolism (E), and coenzyme transport and metabolism (H) were similar to or higher than that found in D. desulfuricans ATCC 27774, and much higher than in L. intracellularis (Figure 4).

Non-supervised orthologous groups (NOG) classification of genes in the genomes of ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31 and reference organisms.
‘Ca. Desulfovibrio trichonymphae’ shared 906 CDSs with D. desulfuricans ATCC 27774, while 176 and 1,450 CDSs were unique in the former and the latter, respectively (Supplementary Figure S6). Among them, genes for superoxide dismutase and cytochrome bd are missing in the ‘Ca. Desulfovibrio trichonymphae’ genome, and genes coding for dye-decolorizing peroxidase, catalase, and rubredoxin-oxygen oxidoreductase are pseudogenized (Supplementary Table S6). Thus, the ability to detoxify oxygen is compromised in ‘Ca. Desulfovibrio trichonymphae’, and the bacterium cannot use oxygen as an electron acceptor unlike certain desulfovibrios (Kuhnigk et al., 1996; Cypionka, 2000). This is in contrast to D. desulfuricans ATCC 27774, which can grow even at 18% O2 (Lobo et al., 2007).
Lateral gene transfer (LGT)
Genes of ‘Ca. Desulfovibrio trichonymphae’ showing the highest sequence similarity to those of non-desulfovibrio bacteria are listed in Supplementary Table S7. Among them, the aromatic amino acid permease AroP was phylogenetically closest to those of ‘Ca. Endomicrobium trichonymphae’ and ‘Ca. Endomicrobium sp.’ HsTcC-Em16 (Supplementary Figure S7A). The codon adaptation indices (CAI) of aroP were 0.69 and 0.73 in ‘Ca. Desulfovibrio trichonymphae’ and ‘Ca. Endomicrobium trichonymphae’, respectively. These values were within the range of 95% confidence intervals (0.65–0.79 and 0.69–0.82, respectively; Supplementary Table S7). These indicate that an LGT of aroP has occurred between the ancestor of ‘Ca. Desulfovibrio trichonymphae’ and the common ancestor of the endosymbiotic Endomicrobium species, and that the LGT was not a very recent event. The putative malate/succinate antiporter DcuA, which plays a crucial role in fumarate respiration (Ullmann et al., 2000), and the fumarate reductase subunits were probably acquired through LGT (Supplementary Table S7 and Supplementary Figures S7B and C).
Discussion
The present study revealed that ‘Ca. Desulfovibrio trichonymphae’ retains the complex system required to oxidize H2 by sulfate and/or fumarate in spite of its reduced genome size. In contrast, the bacterium has lost the ability to utilize other electron donors common in desulfovibrios, such as lactate, formate, and ethanol. The corruption of the repressor gene rex suggested that ‘Ca. Desulfovibrio trichonymphae’ constitutively expresses genes for sulfate respiration. Since the protist host and the co-inhabiting ‘Ca. Endomicrobium trichonymphae’ most probably generate H2 during the fermentation of sugars (Yamin, 1980; Odelson and Breznak, 1985; Hongoh et al., 2008a; Zheng et al., 2016), we suggest that interspecies H2 transfer is one of the driving forces for the evolution of this tripartite symbiosis. Consistently, ‘Ca. Desulfovibrio trichonymphae’ cells are localized immediately adjacent to hydrogenosome-like compartments (Figure 1; Sato et al., 2009), organelles that produce H2 (Müller et al. 2012).
Although the H2 partial pressure in the protist-inhabiting portion (paunch) of agarose-embedded termite guts is extremely high (for example, 15–30 kPa in Reticulitermes santonensis), the H2-emission rate of living termites is 30 to 50 fold lower (Ebert and Brune, 1997; Pester and Brune, 2007). In addition, the H2 partial pressure steeply decreases toward the peripheral gut region (Ebert and Brune, 1997; Pester and Brune, 2007). These indicate that H2 is rapidly removed by H2-oxidizers, especially in the gut of living termites, and imply that competition for H2 can occur among those microbes. Indeed, exogenously-supplied H2 greatly enhances the methanogenic activity of the termites Zootermopsis angusticolis and Reticulitermes flavipes, both of which harbour methanogens that produce CH4 from H2 and CO2 in their gut (Messer and Lee, 1989; Ebert and Brune, 1997). Therefore, it is tempting to infer that an ancestor of ‘Ca. Desulfovibrio trichonymphae’ might have colonized the surface of a Trichonympha cell for an abundant H2 supply, and subsequently evolved into a vertically transmitted, almost intracellular symbiont. The small pore may be necessary for sulfate uptake from the outside of the host protist cell. The colonization on the Trichonympha cells should also have eliminated the cost and need for its own motility that is otherwise required to keep the bacterium at nutritionally optimal sites in the gut and to prevent washout from the gut.
The removal of H2 by ‘Ca. Desulfovibrio trichonymphae’, in turn, likely benefits the protist host and the co-inhabiting ‘Ca. Endomicrobium trichonymphae’ by decreasing the inhibitory effect of H2 against their fermentation processes. The fact that all the T. agilis cells in the R. speratus gut harbour ‘Ca. Desulfovibrio trichonymphae’ strongly suggests that they have a mutualistic relationship. On the other hand, certain Trichonympha species do not possess Desulfovibrio symbionts (Strassert et al., 2012). Other endo- and/or ectosymbionts might substitute the role of ‘Ca. Desulfovibrio trichonymphae’. Otherwise, since H2 diffuses rapidly in the gut environment, a portion of the gut protist community might not need to house H2-oxidizers if there are enough H2-oxidizing activities in total in the gut. Thus, the need for cellular association between a protist and H2-oxidizers might also depend on the total abundance of H2-oxidizers in the gut.
Trichonympha-associated Desulfovibrio phylotypes are not monophyletic (Supplementary Figure S8), implying that independent acquisitions of Desulfovibrio symbionts by Trichonympha protists have occurred (Sato et al., 2009; Strassert et al., 2012; Ikeda-Ohtsubo et al., 2016). Trichonympha collaris in the gut of Zootermopsis nevadensis harbours rod-shaped Desulfovibrio ectosymbionts, which are laterally attached to the host cell surface (Ikeda-Ohtsubo et al., 2016). The Desulfovibrio ectosymbionts of Trichonympha globulosa are held by invaginations much deeper than those of T. collaris (Ikeda-Ohtsubo et al., 2016; Strassert et al., 2012). These might be in intermediate stages in the evolution to the nearly intracellular symbiont like ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31. Since the concentration of sulfate in the termite gut is not high (for example 0.00–0.01 mM in R. speratus and 0.09–0.33 mM in H. sjostedti) (Sato et al., 2009), the supply of malate from the host cytoplasm for fumarate respiration may be another driving force for the symbioses between these Trichonympha and Desulfovibrio species.
Interestingly, T. collaris harbours a third symbiont, ‘Candidatus Adiutrix intracellularis’, in addition to an Endomicrobium endosymbiont and the Desulfovibrio ectosymbiont (Ikeda-Ohtsubo et al., 2016). ‘Ca. Adiutrix intracellularis’ is an intracellular symbiont, which belongs to the ‘Rs-K70 group’, a deeply branching clade in the class Deltaproteobacteria. A draft genome analysis of this bacterium suggested that it produces acetate from H2 and CO2 (Ikeda-Ohtsubo et al., 2016). Transcription of the gene coding for a key enzyme, hydrogenase-linked formate dehydrogenase, was detected in situ (Rosenthal et al., 2013). Interrelationships among these three bacterial symbionts are unknown. Hydrogen-oxidizing activity has been experimentally demonstrated in bacterial symbionts of other termite-gut protists. Eucomonympha sp. in the gut of H. sjostedti harbours an intracellular rod-shaped bacterium, ‘Candidatus Treponema intracellularis’ (order Spirochaetales), which also produces acetate from H2 and CO2 (Ohkuma et al., 2015). ‘Candidatus Azobacteroides pseudotrichonymphae’ (order Bacteroidales), an intracellular symbiont of the protist Pseudotrichonympha grassii in the gut of the termite Coptotermes formosanus, exhibited a strong H2-uptake activity (Inoue et al., 2007; Hongoh et al., 2008b).
The ability to synthesize amino acids and cofactors is, in part, complementary between ‘Ca. Desulfovibrio trichonymphae’ and ‘Ca. Endomicrobium trichonymphae’ (Supplementary Tables S4 and S5). Given that the termite host feeds on nitrogen-poor wood materials, biosynthesis and provision of essential nitrogenous compounds are critically important for the termite, and probably also for the protists (Odelson and Breznak, 1985). In addition, provision of cobalamin from ‘Ca. Desulfovibrio trichonymphae’ to ‘Ca. Endomicrobium trichonymphae’ should be important because the latter has a cobalamin-dependent methionine synthase and a cobalamin-dependent ribonucleotide reductase (Hongoh et al., 2008a). The synthesized methionine, in turn, can be provided through the host cytoplasm to ‘Ca. Desulfovibrio trichonymphae’, which possesses the transporter MetINQ. Whereas ‘Ca. Desulfovibrio trichonymphae’ probably takes up amino acids, including methionine, also from the gut lumen through the small pore, provision of nitrogen sources from the host cytoplasm without competition seems to be beneficial to the bacterium. Thus, the complementary supply of nitrogenous compounds may be another driving force for the evolution of this symbiosis. It remains unknown how such compounds are released to the host cytoplasm from these symbiotic bacteria; the protist host may digest the symbionts like exogenously supplied bacterial cells (Odelson and Breznak, 1985).
Because the gene for ammonium transporter is pseudogenized in ‘Ca. Endomicrobium trichonymphae’ (Hongoh et al., 2008a) and absent in ‘Ca. Desulfovibrio trichonymphae’, amino acids should be their primary nitrogen sources. Interestingly, the aromatic amino acid transporter aroP gene has obviously been laterally transferred between the ancestors of ‘Ca. Desulfovibrio trichonymphae’ and the endosymbiotic endomicrobia. Genes required for the fumarate respiration of ‘Ca. Desulfovibrio trichonymphae’ may have also been laterally acquired from other bacterial lineages (Supplementary Table S7). It has been suggested that ‘Ca. Adiutrix intracellularis’ laterally acquired genes required for its reductive acetogenesis (Ikeda-Ohtsubo et al., 2016). Thus, LGT plays important roles in the symbiotic system in the termite gut.
In conclusion, our analyses of the complete genomes of the two symbionts ‘Ca. Desulfovibrio trichonymphae’ and ‘Ca. Endomicrobium trichonymphae’ unveiled an elaborate mutualism within the protist cell. A schematic tripartite symbiosis is depicted in Figure 5. The gut protists phagocytose wood particles, hydrolyze the cellulose and hemicellulose to monosaccharides, and ferment the monosaccharides to acetate, CO2 and H2 (Odelson and Breznak, 1985; Yamin, 1980). A portion of the monosaccharides are imported by ‘Ca. Endomicrobium trichonymphae’ and fermented to acetate, CO2, ethanol, and H2 (Hongoh et al., 2008a). The produced acetate is the main carbon and energy source of the termite host (Brune, 2014). ‘Ca. Desulfovibrio trichonymphae’ takes up the generated H2 as the energy source and acetate and CO2 as the carbon sources. Malate, produced during the fermentation process of the protist, enables ‘Ca. Desulfovibrio trichonymphae’ to oxidize H2 even when sulfate is unavailable. The removal of H2 should promote the fermentation processes of the protist and ‘Ca. Endomicrobium trichonymphae’. The protist provides these bacteria with the metabolites and the habitat, and in turn, obtains various amino acids and cofactors. Future studies of the functions of the uncultivable protist host are needed to fill the picture describing this complex symbiotic system.

Proposed tripartite symbiotic relationship among ‘Ca. Desulfovibrio trichonymphae’ phylotype Rs-N31, ‘Ca. Endomicrobium trichonymphae’ phylotype Rs-D17, and Trichonympha agilis.
Accession codes
References
Badziong W, Ditter B, Thauer RK . (1979). Acetate and carbon dioxide assimilation by Desulfovibrio vulgaris (Marburg), growing on hydrogen and sulfate as sole energy source. Arch Microbiol 123: 301–305.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477.
Brune A . (2014). Symbiotic digestion of lignocellulose in termite guts. Nat Rev Microbiol 12: 168–180.
Christensen GA, Zane GM, Kazakov AE, Li X, Rodionov DA, Novichkov PS et al. (2015). Rex (encoded by DVU_0916) in Desulfovibrio vulgaris Hildenborough is a repressor of sulfate adenylyl transferase and is regulated by NADH. J Bacteriol 197: 29–39.
Cypionka H . (2000). Oxygen respiration by Desulfovibrio species. Annu Rev Microbiol 54: 827–848.
Do TH, Nguyen TT, Nguyen TN, Le QG, Nguyen C, Kimura K et al. (2014). Mining biomass-degrading genes through Illumina-based de novo sequencing and metagenomic analysis of free-living bacteria in the gut of the lower termite Coptotermes gestroi harvested in Vietnam. J Biosci Bioeng 118: 665–671.
Donmez N, Brudno M . (2013). SCARPA: scaffolding reads with practical algorithms. Bioinformatics 29: 428–434.
Ebert A, Brune A . (1997). Hydrogen concentration profiles at the oxic-anoxic interface: a microsensor study of the hindgut of the wood-feeding lower termite Reticulitermes flavipes (Kollar). Appl Environ Microbiol 63: 4039–4046.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Greene EA, Hubert C, Nemati M, Jenneman GE, Voordouw G . (2003). Nitrite reductase activity of sulphate-reducing bacteria prevents their inhibition by nitrate-reducing, sulphide-oxidizing bacteria. Environ Microbiol 5: 607–617.
Grein F, Ramos AR, Venceslau SS, Pereira IAC . (2013). Unifying concepts in anaerobic respiration: insights from dissimilatory sulfur metabolism. Biochim Biophys Acta 1827: 145–160.
Grissa I, Vergnaud G, Pourcel C . (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35: W52–W57.
Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF et al. (2004). The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol 22: 554–559.
Hongoh Y . (2011). Toward the functional analysis of uncultivable, symbiotic microorganisms in the termite gut. Cell Mol Life Sci 68: 1311–1325.
Hongoh Y, Sharma VK, Prakash T, Noda S, Taylor TD, Kudo T et al. (2008a). Complete genome of the uncultured Termite Group 1 bacteria in a single host protist cell. Proc Natl Acad Sci USA 105: 5555–5560.
Hongoh Y, Sharma VK, Prakash T, Noda S, Toh H, Taylor TD et al. (2008b). Genome of an endosymbiont coupling N2 fixation to cellulolysis within protist cells in termite gut. Science 322: 1108–1109.
Ikeda-Ohtsubo W, Strassert JFH, Köhler T, Mikaelyan A, Gregor I, McHardy AC et al. (2016). ‘Candidatus Adiutrix intracellularis’, an endosymbiont of termite gut flagellates, is the first representative of a deep-branching clade of Deltaproteobacteria and a putative homoacetogen. Environ Microbiol 18: 2548–2564.
Inoue JI, Saita K, Kudo T, Ui S, Ohkuma M . (2007). Hydrogen production by termite gut protists: characterization of iron hydrogenases of parabasalian symbionts of the termite Coptotermes formosanus. Eukaryot Cell 6: 1925–1932.
Keller KL, Wall JD . (2011). Genetics and molecular biology of the electron flow for sulfate respiration in Desulfovibrio. Front Microbiol 2: 1–17.
Kuhnigk T, Branke J, Krekeler D, Cypionka H, König H . (1996). A feasible role of sulfate-reducing bacteria in the termite gut. Syst Appl Microbiol 19: 139–149.
Lobo SAL, Melo AMP, Carita JN, Teixeira M, Saraiva LM . (2007). The anaerobe Desulfovibrio desulfuricans ATCC 27774 grows at nearly atmospheric oxygen levels. FEBS Lett 581: 433–436.
Martin M . (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17: 10–12.
Mcorist S, Gebhart CJ, Boid R, Barns SM . (1995). Characterization of Lawsonia intracellularis gen. nov., sp. nov., the obligately intracellular bacterium of porcine proliferative enteropathy. Int J Syst Bacteriol 45: 820–825.
Messer AC, Lee MJ . (1989). Effect of chemical treatments on methane emission by the hindgut microbiota in the termite Zootermopsis angusticollis. Microbial Ecol 18: 275–284.
Meyer B, Kuehl JV, Price MN, Ray J, Deutschbauer AM, Arkin AP et al. (2014). The energy-conserving electron transfer system used by Desulfovibrio alaskensis strain G20 during pyruvate fermentation involves reduction of endogenously formed fumarate and cytoplasmic and membrane-bound complexes, Hdr-Flox and Rnf. Environ Microbiol 16: 3463–3486.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M . (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–W185.
Müller M, Mentel M, van Hellemond JJ, Henze K, Woehle C, Gould SB et al. (2012). Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol Mol Biol R 76: 444–495.
Novichkov PS, Kazakov AE, Ravcheev DA, Leyn SA, Kovaleva GY, Sutormin RA et al. (2013). RegPrecise 3.0-a resource for genome-scale exploration of transcriptional regulation in bacteria. BMC Genom 14: 745.
Odelson DA, Breznak JA . (1985). Nutrition and growth characteristics of Trichomitopsis termopsidis, a cellulolytic protozoan from termites. Appl Environ Microb 49: 614–621.
Odelson DA, Breznak JA . (1985). Cellulase and other polymer-hydrolyzing activities of Trichomitopsis termopsidis, a symbiotic protozoan from termites. Appl Environ Microb 49: 622–626.
Ohkuma M, Noda S, Hattori S, Iida T, Yuki M, Starns D et al. (2015). Acetogenesis from H2 plus CO2 and nitrogen fixation by an endosymbiotic spirochete of a termite-gut cellulolytic protist. Proc Natl Acad Sci USA 112: 10224–10230.
Ohkuma M, Sato T, Noda S, Ui S, Kudo T, Hongoh Y . (2007). The candidate phylum 'Termite Group 1' of bacteria: phylogenetic diversity, distribution, and endosymbiont members of various gut flagellated protists. FEMS Microbiol Ecol 60: 467–476.
Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS . (2011). A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front Microbiol 2: 1–22.
Pester M, Brune A . (2007). Hydrogen is the central free intermediate during lignocellulose degradation by termite gut symbionts. ISME J 1: 551–565.
Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J et al. (2014). eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res 42: D231–D239.
Puigbo P, Bravo IG, Garcia-Vallve S . (2008). CAIcal: a combined set of tools to assess codon usage adaptation. Biol Direct 3: 38.
Ramos AR, Grein F, Oliveira GP, Venceslau SS, Keller KL, Wall JD et al. (2015). The FlxABCD-HdrABC proteins correspond to a novel NADH dehydrogenase/heterodisulfide reductase widespread in anaerobic bacteria and involved in ethanol metabolism in Desulfovibrio vulgaris Hildenborough. Environ Microbiol 17: 2288–2305.
Ravcheev DA, Li XQ, Latif H, Zengler K, Leyn SA, Korostelev YD et al. (2012). Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J Bacteriol 194: 1145–1157.
Rosenthal AZ, Zhang XN, Lucey KS, Ottesen EA, Trivedi V, Choi HMT et al. (2013). Localizing transcripts to single cells suggests an important role of uncultured deltaproteobacteria in the termite gut hydrogen economy. Proc Natl Acad Sci USA 110: 16163–16168.
Santana M, Crasnier-Mednansky M . (2006). The adaptive genome of Desulfovibrio vulgaris Hildenborough. FEMS Microbiol Lett 260: 127–133.
Sato T, Hongoh Y, Noda S, Hattori S, Ui S, Ohkuma M . (2009). Candidatus Desulfovibrio trichonymphae, a novel intracellular symbiont of the flagellate Trichonympha agilis in termite gut. Environ Microbiol 11: 1007–1015.
Sato T, Kuwahara H, Fujita K, Noda S, Kihara K, Yamada A et al. (2014). Intranuclear verrucomicrobial symbionts and evidence of lateral gene transfer to the host protist in the termite gut. ISME J 8: 1008–1019.
Schmieder R, Edwards R . (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27: 863–864.
Stingl U, Radek R, Yang H, Brune A . (2005). ‘Endomicrobia’: cytoplasmic symbionts of termite gut protozoa form a separate phylum of prokaryotes. Appl Environ Microb 71: 1473–1479.
Strassert JFH, Köhler T, Wienemann THG, Ikeda-Ohtsubo W, Faivre N, Franckenberg S et al. (2012). ‘Candidatus Ancillula trichonymphae’, a novel lineage of endosymbiotic Actinobacteria in termite gut flagellates of the genus Trichonympha. Environ Microbiol 14: 3259–3270.
Talavera G, Castresana J . (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56: 564–577.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S . (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729.
Tartar A, Wheeler MM, Zhou XG, Coy MR, Boucias DG, Scharf ME . (2009). Parallel metatranscriptome analyses of host and symbiont gene expression in the gut of the termite Reticulitermes flavipes. Biotechnol Biofuels 2: 25.
Tokuda G, Tsuboi Y, Kihara K, Saitou S, Moriya S, Lo N et al. (2014). Metabolomic profiling of 13C-labelled cellulose digestion in a lower termite: insights into gut symbiont function. Proc R Soc B-Biol Sci 281: 20140990.
Trager W . (1934). The cultivation of a cellulose-digesting flagellate, Trichomonas termopsidis, and of certain other termite protozoa. Biol Bull 66: 182–190.
Ullmann R, Gross R, Simon J, Unden G, Kroger A . (2000). Transport of C4-dicarboxylates in Wolinella succinogenes. J Bacteriol 182: 5757–5764.
Winnen B, Hvorup RN, Saier MH . (2003). The tripartite tricarboxylate transporter (TTT) family. Res Microbiol 154: 457–465.
Yamin MA . (1980). Cellulose metabolism by the termite flagellate Trichomitopsis termopsidis. Appl Environ Microb 39: 859–863.
Zheng H, Dietrich C, Radek R, Brune A . (2016). Endomicrobium proavitum, the first isolate of Endomicrobia class. nov. (phylum Elusimicrobia – an ultramicrobacterium with an unusual cell cycle that fixes nitrogen with a Group IV nitrogenase. Environ Microbiol 18: 191–204.
Acknowledgements
We thank Rie Ryusui and Takehiko Itoh for helpful advice on DNA sequencing on an Illumina MiSeq. Sanger sequencing was done in RIKEN BSI, and TEM was assisted by Keiko Ikeda in the Biomaterial Analysis Center in the Tokyo Institute of Technology. This study was financially supported by the NEXT program (GS009) to YH from JSPS, MEXT KAKENHI to HK (15K18604), YH (26650158), and to YH and MO (23117003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Kuwahara, H., Yuki, M., Izawa, K. et al. Genome of ‘Ca. Desulfovibrio trichonymphae’, an H2-oxidizing bacterium in a tripartite symbiotic system within a protist cell in the termite gut. ISME J 11, 766–776 (2017). https://doi.org/10.1038/ismej.2016.143
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2016.143
This article is cited by
-
Functional similarity, despite taxonomical divergence in the millipede gut microbiota, points to a common trophic strategy
Microbiome (2024)
-
Diet Influences the Gut Microbial Diversity and Olfactory Preference of the German Cockroach Blattella germanica
Current Microbiology (2023)
-
The functional evolution of termite gut microbiota
Microbiome (2022)
-
Agarose gel microcapsules enable easy-to-prepare, picolitre-scale, single-cell genomics, yielding high-coverage genome sequences
Scientific Reports (2022)
-
Potential of termite gut microbiota for biomethanation of lignocellulosic wastes: current status and future perspectives
Reviews in Environmental Science and Bio/Technology (2021)
