
Abstract
Submarine hydrothermal vents perturb the deep-ocean microbiome by injecting reduced chemical species into the water column that act as an energy source for chemosynthetic organisms. These systems thus provide excellent natural laboratories for studying the response of microbial communities to shifts in marine geochemistry. The present study explores the processes that regulate coupled microbial-geochemical dynamics in hydrothermal plumes by means of a novel mathematical model, which combines thermodynamics, growth and reaction kinetics, and transport processes derived from a fluid dynamics model. Simulations of a plume located in the ABE vent field of the Lau basin were able to reproduce metagenomic observations well and demonstrated that the magnitude of primary production and rate of autotrophic growth are largely regulated by the energetics of metabolisms and the availability of electron donors, as opposed to kinetic parameters. Ambient seawater was the dominant source of microbes to the plume and sulphur oxidisers constituted almost 90% of the modelled community in the neutrally-buoyant plume. Data from drifters deployed in the region allowed the different time scales of metabolisms to be cast in a spatial context, which demonstrated spatial succession in the microbial community. While growth was shown to occur over distances of tens of kilometers, microbes persisted over hundreds of kilometers. Given that high-temperature hydrothermal systems are found less than 100 km apart on average, plumes may act as important vectors between different vent fields and other environments that are hospitable to similar organisms, such as oil spills and oxygen minimum zones.
Similar content being viewed by others


Introduction
Microbial life is abundant throughout the oceans and has a key role in modulating global geochemical cycles (Falkowski et al., 2008). The advent of advanced molecular tools has provided an invaluable means of studying spatial and temporal variations in the phylogenetic structure and geochemical function of these marine microbial communities (Gilbert and Dupont, 2011). Such insights are critical for predicting the future of the oceans given that anthropogenic perturbations, like climate change and coastal eutrophication, are causing marine ecosystems to deviate from their natural states with uncertain consequences (Nogales et al., 2013). While mathematical models are often used to make such predictions (for example, IPCC, 2013)—as well as to elucidate underlying mechanisms and to quantify processes—a current challenge is incorporating data from cutting-edge molecular tools into biogeochemical models.
The present communication focuses on predicting the response of microbial communities to geochemical perturbations in the deep ocean, which remains an enigmatic environment despite constituting the largest ecosystem on Earth (Arístegui et al., 2009). In contrast to sunlit surface waters, primary production in the aphotic zone is typically energy limited. Deep-sea autotrophs generate energy by mediating redox reactions and, given that the oceans are largely oxygenated, growth is chiefly regulated by the availability of electron donors (Jannasch and Wirsen, 1979). Hydrothermal systems, located along mid-ocean ridges, are a major source of these electron donors to the deep ocean. In these regions, hot fluids rich in reduced chemical species rise from beneath the seafloor and are injected into the cold, oxic water column (Tivey, 2004), stimulating microbial growth as a result (Winn et al., 1986). These natural geochemical perturbations thus provide an excellent opportunity for studying marine microbial dynamics. Chemosynthetic organisms support extensive biological communities at hydrothermal vents (Grassle, 1986). Nevertheless, their impact is not confined to the vent locale as hydrothermal plumes distribute material throughout the oceans as they rise, spread laterally, and become entrained in deep circulation (Tagliabue et al., 2010). Plumes may thus act as vectors, linking microbial communities in various marine environments (Dick et al., 2013). While recent metagenomic and metatranscriptomic studies have provided much-needed insight into those microbes present in plumes as well as their metabolisms (Baker et al., 2012; Lesniewski et al., 2012; Anantharaman et al., 2013; Baker et al., 2013; Marshall and Morris, 2013; Li et al., 2014; Sheik et al., in press), fundamental questions remain regarding the ecology and dynamics of these communities. Both near-vent and pelagic communities have been invoked as the origin of plume-dwelling microbes (Winn et al., 1986; Lesniewski et al., 2012), but how these potential sources interact with plume physics, vent chemistry and microbial growth to shape plume communities remains unclear.
Given that energy drives deep-sea autotrophy, thermodynamic mixing models have been used extensively over the past 20 years to estimate primary production in chimney walls, plumes and low-temperature diffuse flow regions of submarine hydrothermal systems (for example, Shock et al., 1995; McCollom and Shock, 1997; McCollom, 2000, 2007; Amend et al., 2011; Anantharaman et al., 2013; Boettger et al., 2013; Anantharaman et al., 2014; Nakamura and Takai, 2014). These models calculate the amount of energy that is potentially available through a wide range of pathways, thus predicting the prevalence of these metabolisms. More recently, a reactive-transport model (RTM) of an ideal hydrothermal chimney was developed that not only considers thermodynamics, like previous models, but also explicitly incorporates reaction kinetics, allowing processes that occur within chimney walls to be spatially resolved (LaRowe et al., 2014). These studies have provided great insight into the microbial metabolisms that are potentially active within hydrothermal systems, but are largely theoretical and do not make detailed, site-specific comparisons with microbial data. A recent study has sought to address this issue by developing a metastable equilibrium model that was fit directly to phylum-level microbial data from a hydrothermal spring in Yellowstone National Park (Dick and Shock, 2013). However, while this approach is able to reproduce observations well, it is unable to forge links between the microbial community and geochemical dynamics since metabolic function cannot be assigned based on phyla and the model does not explicitly include biogeochemical processes.
Recently, a modelling framework was advanced that couples marine microbiology and geochemistry by directly linking microbial growth, reaction kinetics and thermodynamics (Reed et al., 2014). The microbial community is divided according to metabolism and these sub-populations are tracked using associated functional genes. Previously, this method has been successfully applied to study the biogeochemistry of oxygen minimum zones using qPCR to quantify absolute functional gene abundance (Reed et al., 2014). In contrast, here we employ the approach in the context of a spatially (and temporally) resolved hydrothermal plume with the model directly driven by and validated with metagenomic data derived from samples taken in the plume and from the surrounding water column. The model integrates physico-biogeochemical processes associated with hydrothermal plumes into a coherent whole and is applied to a site in the Lau basin, a back-arc basin located in the west of the South Pacific Ocean. The overarching goal of the present study is to explain the structure of microbial communities within hydrothermal plumes, while identifying the environmental factors that govern the geochemical potential of these communities. First, we consider the role of physics in shaping the microbial community in a hydrothermal plume. Then, we introduce a subset of seven chemolithoautotrophic functional groups (that is, seven metabolic pathways) and demonstrate biogeochemical processes in action. Next, an additional pathway, hydrogen oxidation, is incorporated to explore a more complex scenario involving metabolic versatility. Finally, we use the results of these simulations to consider the broader impact of plumes on deep-sea biogeochemistry.
Materials and methods
Physico-biogeochemical model
A RTM of a hydrothermal plume was developed that considers the distributions and fates of chemical and biological species associated with various chemolithoautotrophic metabolisms. Microbial populations are modelled using a gene-centric approach, whereby organisms that mediate different metabolic pathways are tracked using abundances of associated functional genes (Reed et al., 2014). For example, a state variable for the amoA gene represents the abundance of ammonia oxidising microbes. In addition, there are also state variables for metabolic reactants (for example, oxygen and ammonia) and products (for example, nitrite). Each sub-population is associated with one metabolism with the exception of sulphur oxidation. The dominant sulphur oxidiser in the region, SUP05 (Sheik et al., in press; K Anantharaman, personal communication), possesses all of the genes necessary to completely oxidise various reduced sulphur species to sulphate (Anantharaman et al., 2013). Consequently, these metabolisms are combined into a single group. All metabolisms that are considered by the model are presented in Supplementary Table S1.
Within the RTM, microbial growth is described by means of Michaelis-Menten kinetics for both electron donors and acceptors (see Supplementary Material). Uptake of enzymatic substrates is linked to growth using an empirical relationship that expresses growth yield as a function of energy of the metabolism (Roden and Jin, 2011). Mortality is modelled using a first-order term for gene abundance. These biogeochemical processes—growth, death, consumption and production—are then cast into a physical transport framework. Parameters pertaining to these processes are given in Supplementary Table S2.
While the Roden and Jin empirical model (RJM, hereafter) that relates biomass yield and metabolic energy yield (that is, Roden and Jin, 2011) provides an invaluable first-order estimate of this relationship, there are a number of issues that must be borne in mind when employing this approach. First, the RJM is largely derived from organic metabolisms—with the exception of aerobic hydrogen oxidation—whereas we consider inorganic reactions here. Nevertheless, to our knowledge there is currently no equivalent model exclusively for inorganic metabolisms. Next, the model may overestimate biomass yield, as it derived from experiments performed under optimal conditions. Consequently, the model does not account for transient environmental conditions or other natural pressures. Also, since the studies used to derive the relationship did not typically provide detailed information about the concentrations of the reactants and products—data that are required to calculate metabolic energy yields—these concentrations were estimated from typical culture experiments. Finally, the model does not consider the impact of maintenance energy of biomass production, which could be potentially important in natural settings. Nevertheless, despite these caveats, the model remains a useful tool.
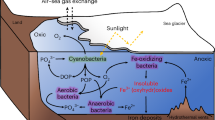
Hot fluid emanating from hydrothermal vents is less dense than seawater and thus ascends as it is injected into the cold water column, forming a plume as a result. Ambient seawater is entrained as the plume rises, reducing its buoyancy and vertical velocity until a neutral buoyancy is ultimately achieved (Helfrich and Speer, 1995; McDuff, 1995). Momentum carries rising plume water beyond this point, before gravity returns it to the depth of neutral buoyancy and spreads laterally (Jiang and Breier, 2014). With this structure in mind, the model’s physical domain is divided into two connected regions: the rising plume and the neutrally-buoyant plume.
The rising plume is spatially resolved along a 1D domain that is oriented vertically, parallel to flow (Figure 1). Rise velocity, turbulent mixing and the geometry of the rising plume are prescribed from an existing 2D axisymmetric fluid dynamics model (FDM; Jiang and Breier, 2014). In the RTM, the 1D rising plume domain is divided vertically into a series of adjacent, non-overlapping volumes. The height of these volumes is set by the node spacing along the centre axis of the plume from the FDM. The radius of each of these volumes—that is, the edge of the rising plume—is determined as the first point in the FDM where the lateral velocity is towards the centre of the plume (that is, where entrainment begins). The efflux of water at the top of each volume is typically greater than the influx at the base of the volume and, therefore, neighbouring seawater is entrained into the rising plume to conserve volume. In the RTM, rise velocities and turbulent mixing coefficients are an average of all FDM nodes that fall within each volume.

A schematic of the model domain showing the shape of the rising plume, entrainment of ambient seawater, and the export of a water parcel to the neutrally-buoyant plume.
Parameters for the rising plume region of the RTM were generated by the FDM during simulation rke7_0 (Jiang and Breier, 2014), which represents a steady-state plume emanating from vent A1 located in the ABE hydrothermal field (20°45.672883’ S 176°11.434418’ W, depth 2140 m). Within the FDM, a k-ɛ model is used to represent turbulence, and mixing coefficients for the RTM are calculated from the turbulent viscosities output from the FDM, assuming a turbulent Schmidt number of 0.7. For simulation rke7_0, ambient seawater temperature was prescribed as 2.2 °C at the base of the FDM domain and 2.3663 °C at the top of the domain (that is, 400 m above the seafloor), while ambient salinity was set as 34.65 resulting in a buoyancy frequency 0.000473 s−1 (Jiang and Breier, 2014). The chimney diameter and height were set at 0.14 m and 2 m, respectively, and hydrothermal fluid was expelled at 0.2 ms−1, with a temperature of 309 °C. The resulting plume achieved a maximum height of 201 m. For thermodynamic calculations, the pressure is assumed to be a constant 210 bar.
Unlike the spatially resolved rising plume, the neutrally-buoyant plume is simulated in the RTM by tracking a unit volume of water exported from the rising plume over time as it spreads laterally. No additional dilution is assumed to occur in the neutrally-buoyant plume, because mixing is substantially lower than in the rising plume (McCollom, 2000), and we assume no feedback to the rising plume.
To run the model, values for the chemical and biological state variables must be prescribed at the vent orifice and in the surrounding seawater. The chemistry of nearby seawater is defined to represent 'typical' deep-sea water. Initial plume chemistry is prescribed using an existing reaction-path model (Breier et al., 2012; Anantharaman et al., 2014), which accounts for the rapid abiotic precipitation of minerals and chemical speciation in the vicinity of the chimney that results from the mixing of vent fluid (Mottl et al., 2011; Flores et al., 2012) and background seawater (Supplementary Table S3); details of this model for the A1 vent are described in Anantharaman et al. (2014).
Output of simulations describes the distribution of state variables over space (in the rising plume) and time (in the neutrally-buoyant plume). The RTM is coded in R (R Development Core Team, 2006) using the tran.volume.1D function of the ReacTran package to calculate transport terms (Soetaert and Meysman, 2012), while thermodynamic calculations are undertaken using the CHNOSZ package (Dick, 2008). Solutions are achieved by running the RTM to steady state in the rising plume using the vode numerical integrator from the deSolve package (Soetaert et al., 2010). The model code is available at www.earth.lsa.umich.edu/geomicrobiology/Lau.html.
Metagenomic data
Microbial data used to drive and validate the model were collected from the ABE hydrothermal vent field, Lau basin, during cruises TN235 and TN236 aboard the R/V Thomas G Thompson in May–July 2009 (Supplementary Table S4; Anantharaman et al., 2014). Sampling was undertaken using a suspended-particle rosette multi-sampler (Breier et al., 2009) with 0.8 μm filters. DNA extracted from ¼ of a filter, as described by Dick and Tebo (2010), was amplified using multiple displacement amplification before undergoing shotgun sequencing (Anantharaman et al., 2014). Sequences can be accessed from the NCBI Sequence Read Archive using accession numbers SRX511269, SRX511304 and SRX511275 (see Supplementary Table S4).
Samples were collected by precision navigation using the remotely operated vehicle (ROV) Jason in dynamic positioning mode. Jason's position relative to the seafloor and target vent was measured in real time by long baseline acoustic navigation, supplementing the depth (pressure) sensor and altimeter on the vehicle. Using this navigation system, we found the orifice of our target vent, ABE, and sampled directly at the base of the plume, which was identified visually using the ROV. Jason was then directed to rise into the plume following its trajectory, which could be tracked visually using the backscatter from the vehicles laser pointers to 60 m above bottom. From there, the ROV then rose directly to the non-buoyant plume height of 200 m. Synoptic particulate samples were taken and the Al/(Al+Fe+Mn) ratio of these particles determined to confirm that sampling occurred within the plume, as reported by Sheik et al. (in press). The field methodology for this ROV-based plume sampling was described in Breier et al. (2009, 2014). The latter study includes a Supplementary video showing this approach.
Custom databases were created for functional genes associated with the modelled metabolic pathways (Supplementary Table S5). These databases were constructed using a combination of COG/Pfam-based functional annotations, identification of metabolic genes in the Lau metagenome (Anantharaman et al., 2014), and reference genes identified from NCBI GenBank. These databases are available online at the following URL: www.earth.lsa.umich.edu/geomicrobiology/Lau.html. Raw shotgun sequencing reads were trimmed using the adaptive read trimmer, Sickle (https://github.com/najoshi/sickle). Trimming removed 0.14–2.8% of low quality reads. Trimmed forward and reverse reads from a single sample site were concatenated and BLASTX (Altschul et al., 1990) was performed against the custom databases for individual genes using the following parameters: e value <1e−5, alignment length >60 bp, bit score >50, percent identity >60. The resultant data were normalised for variance in data set size and gene length.
Results and discussion
Entrainment shapes rising plume communities
In the model, microbial populations and chemical species are tracked over a 1D domain representing the rising plume and then over time within the neutrally-buoyant plume (Figure 1). Figure 2 shows rise velocity and average time within the plume as a function of height above the orifice. Vent fluid initially accelerates after it is injected into the water column, as it is less dense than seawater and, therefore, more buoyant (Jiang and Breier, 2014). Seawater is quickly entrained and mixed with this vent fluid near the chimney, reducing its buoyancy and slowing the ascent of the plume (Converse et al., 1984). As a result, the maximum rise velocity (∼70 cm s−1) is achieved just above the orifice. Maximum average time in the rising plume is less than 40 min, which is quite short relative to the doubling time of low-temperature, deep-sea chemolithoautrophs (for example, Martens-Habbena et al., 2009) suggesting minimal growth in this region. Nevertheless, before considering the role that biological and geochemical factors play in shaping plume ecosystems, the model is used to examine the impact of physics.

(a) Rise velocity (gray solid line) and average time that vent fluid has spent within the rising plume (black dotted line). (b) Distribution within the plume of inert tracers of microbial communities that originate from the chimney region (black solid line), demersal layer (pink dot-dashed line) and pelagic seawater (blue dotted line).
As they rise, hydrothermal plumes entrain large quantities of seawater, which contain ambient microbial communities and chemical species. The initial plume community (comprised of microbes near the vent orifice) is thus diluted with neighbouring waters that bear potentially different microbial communities. To quantify the influence of entrainment on the structure of plume communities, a simulation was undertaken in which marker genes were used to track inert microbial populations (that is, no growth or mortality) from three regions: near chimney and vent fluid, ambient seawater, demersal (that is, near-bottom) layer. Results demonstrate that, in the absence of growth and mortality, microbes living upon or around chimney structures that are drawn into the plume at the vent orifice are rapidly diluted to near zero abundances due to entrainment of seawater (Figure 2). These organisms may be thermophilic and would therefore be unable to grow appreciably in the rising plume, meaning their contribution to the microbial community is likely negligible above the initial ∼10 m of the plume. This suggests that vent or chimney communities cannot be invoked as the source of elevated cell counts in rising plumes (for example, Winn et al., 1986).
Demersal microbial communities, sustained by diffuse flow of reduced chemicals emanating from the broader vent region, are assumed to occupy a 10-m layer overlying the seafloor in the simulation presented in Figure 2 (Huber et al., 2003). These microbes are quickly entrained into the plume and reach a maximum abundance at the upper edge of the layer. Beyond the demersal layer, their numbers decline due to entrainment of seawater, as seen previously for chimney communities, although at a slower rate. Indeed, demersal microbes are still found at appreciable abundances at 50 m height above the vent orifice. If these organisms are able to utilise substrates of vent origin at relatively low temperatures and low concentrations, then entrainment effectively primes the plume with the near-bottom community ready for growth. A potential example of this in nature is the gammaproteobacterium SUP05, which is found in diffuse flow regimes yet also thrives in hydrothermal plumes (Huber et al., 2003; Sunamura et al., 2004; Anderson et al., 2012; Lesniewski et al., 2012; Marshall and Morris, 2013).
At the uppermost point of the modelled rising plume, the ratio of vent fluid to ambient seawater is on the order of 1:104, consistent with observations (for example, Lupton et al., 1985). Thus, when considering physical processes alone, pelagic deep-sea microbes are shown to dominate the microbial community over the majority of the plume due to entrainment of ambient seawater (Figure 2). This is in agreement with previous studies of hydrothermal plumes (for example, Lesniewski et al., 2012). Nevertheless, these communities are also influenced by biogeochemical processes, which depend on numerous factors, such as the availability of enzymatic substrates and nutrients, cell abundances and mortality. These processes are discussed below.
Predicting the functional composition of the microbial community
Incorporating microbial communities into the model requires that the abundances of each functional group are specified at the vent orifice and within the surrounding ambient seawater. These values are determined from observations using metagenomic data, which give the relative abundances of pertinent functional genes, and an assumed total abundance of 106 functional genes L−1, reflecting deep-sea cell abundances on the order of 106–107 cell l−1 (for example, Karner et al., 2001). The abundance of microbes in ambient seawater is assumed to increase by 5.5% over the height of the plume at ABE (Karner et al., 2001). Metagenomic data are also available for the microbial community within the neutrally-buoyant plume, at a height of 200 m above the vent orifice, which is used to validate model results through comparison with the community predicted by the model in this region.
Simulations undertaken with active microbial communities demonstrate that there is negligible growth in the rising plume and, therefore, the structure of the microbial community entering the neutrally-buoyant plume is very close to that of ambient seawater (results not shown). The majority of microbial growth thus occurs in the neutrally-buoyant plume, as shown in Figure 3, which follows the temporal development of the microbial community beyond the rising plume. Simulations show that model predictions and observations are in closest agreement after 132 h when all metabolisms are reproduced accurately (Figure 4). All metabolisms exhibit growth over time, although the rates of gene production depend on the availability of the associated electron donor and the energy yield of the reaction (Figure 3b). As a result of these differing rates, there is a marked shift in the relative abundances of genes over time (Figure 3a).

Relative and absolute abundances of functional genes for modelled microbial communities associated with seven different aerobic metabolisms versus time in the neutrally-buoyant plume. (Note that four pathways are consolidated into the sulphur oxidation pathway.) The solid black line shows the baseline scenario and the grey regions show range achieved from a Monte Carlo experiment using growth parameters selected randomly from within ±25% of baseline values assuming a uniform distribution of parameters. Vertical, black dotted link shows the point of closest agreement between observations and predictions. Pink dotted line shows the observed community within the neutrally-buoyant plume, while the blue dotted line shows the observed community of ambient seawater.

A comparison of observed (pink solid) and predicted (pink cross-hatched) relative abundances of functional genes after 132 h within the neutrally-buoyant plume (i.e., the moment of closest agreement). Also plotted are the relative abundances of functional genes in background seawater (blue) and at 1 m above vent orifice (black).
While sulphur oxidising microbes are abundant close to the vent orifice—as vent fluids are replete in sulphur species—dilution with seawater attenuates this population to background levels before marked growth occurs in the neutrally-buoyant plume (Figure 3). Ammonia oxidising bacteria and archaea are abundant in ambient seawater, comprising ∼70% of the microbes considered (Figure 4). Nevertheless, in spite of substantial entrainment of such seawater, they represent only around 10% of the observed population in the neutrally-buoyant plume due to marked growth of sulphur oxidisers. Most of the modelled populations exhibit an exponential growth phase followed by a quasi-plateau in absolute abundances when enzymatic substrate is exhausted. Ultimately, after reaching maximum abundances, these populations decline slowly over time due to mortality and an absence of growth (Figure 3a). The absolute abundance of nitrite oxidisers shows an upward curvature over time, however, because nitrite is produced within the plume (Figure 3b). As the ammonia oxidising community grows, the rate of nitrite production increases. Simultaneously, the population of nitrite oxidisers grows, which also enhances the rate of nitrite oxidation. Note that abundance of ammonia oxidisers is much higher than that of nitrite oxidisers, consistent with previous observations in the Guaymas Basin hydrothermal plume (Baker et al., 2012). ABE is hosted in andesitic basalts and, therefore, vent fluid is relatively low in methane, in contrast to ultramafic systems (McCollom, 2007). This is reflected in the microbial community with few methanotrophs present (Figure 3b).
A Monte Carlo approach was employed to examine the impact of kinetic parameters on model output. Half-saturation constants and maximal growth rates for all metabolisms were varied randomly within ±25% of their original values over 50 simulations, assuming a uniform distribution across the ±25% interval, and the results were consolidated to determine the range of responses (grey region, Figure 3). A random parameter sampling approach was used, because relative abundances for each functional group depend not only upon their own growth parameters, but also on growth parameters for other metabolic pathways. While varying growth kinetics affects the specific trajectory of each population, the general trends remain the same since metabolic energy yield exerts a greater influence over the community structure (Figure 3).
Results presented here suggest that energy availability shapes microbial communities that inhabit hydrothermal systems, in agreement with previous studies (for example, Shock et al., 1995; McCollom and Shock, 1997; McCollom, 2000, 2007; Amend et al., 2011; Boettger et al., 2013; Nakamura and Takai, 2014). Nevertheless, while this principle may be accurate in a broad sense, observations that do not conform to this paradigm hint at unresolved mechanisms and additional complexities. One such example is encountered when considering aerobic hydrogen oxidation and the abundance of associated hydrogenases. As shown for ammonia oxidation, rapid growth of sulphur oxidising microbes overwhelms metabolisms that do not experience appreciable growth in the plume, even if they are abundant in the entrained background community (Figure 4). Vent fluids at ABE are depleted in hydrogen, as ABE is hosted in basaltic andesites, which precludes significant growth within the plume via hydrogen oxidation. One would therefore expect the relative abundance of hydrogenases to be attenuated below background levels in the plume; however, this is not the case (Figure 5). These observations are explored in detail below.

A comparison of observed (pink solid) and predicted (grey solid and cross-hatched) relative abundances of functional genes after 132 h within the neutrally-buoyant plume (i.e., the moment of closest agreement). The cross-hatched grey bar shows simulation results without metabolic versatility (i.e., no hydrogenase production by sulphur oxidisers), while the solid grey bar shows results from a simulation with metabolic versatility implemented. Also plotted are the relative abundances of functional genes in background seawater (blue) and at 1 m above the vent orifice (black).
Insights from model-data discrepancies
Expanding upon the suite of metabolisms used above, aerobic hydrogen oxidation was added to the model (that is, the 'Knallgas' reaction, H2+½O2→H2O), as this has been identified as a potentially important pathway in other hydrothermal systems (for example, McCollom, 2000, 2007; Anantharaman et al., 2013). Compared with those systems, however, fluid emanating from the ABE vent is somewhat depleted in H2 (54–101 μmol kg−1; Flores et al., 2012) suggesting that the relative abundance of hydrogenases—genes associated with hydrogen oxidation—should also be low. Nevertheless, in spite of low hydrogen concentrations, substantial dilution, and marked growth by sulphur oxidisers, the relative abundance of hydrogenases remains above background levels (Figure 5). Consequently, assuming that hydrogenase genes are primarily produced using energy derived from hydrogen oxidation leads to discrepancies between model predictions and observations. These disagreements suggest that important mechanisms for hydrogenase production have been omitted from the model.
A recent metagenomic analysis of the vent fields in the Lau basin revealed that sulphur oxidisers are the most abundant chemolithoautotrophs and that SUP05 dominates this metabolism (K Anantharaman, personal communication; Sheik et al., in press). Some populations of SUP05 are metabolically versatile, possessing genes for all sulphur oxidation pathways (hydrogen sulphide, thiosulphate, elemental sulphur, sulphite), as well as hydrogenases (Anantharaman et al., 2013). Given the prevalence of both sulphur oxidation and SUP05 in the region, one would expect these organisms to contribute towards the abundance of hydrogenases. Assuming that SUP05 constitutes ∼20% of the plume’s sulphur oxidising community and, therefore, contributes towards hydrogenase production, generates model predictions that are in close agreement with observations (Figure 5). Metagenomic analysis reveals that the sulphur oxidising community in ambient seawater is 10% SUP05 and 27% SUP05 at 1 m above the vent, which presumably reflects the near-vent demersal community (for example, from the diffuse flow region, biofilms on chimney structures) because vent fluids are too hot to host a native microbial community. Given that the plume community is a mixture of demersal and pelagic communities, these data suggest that assuming the sulphur oxidising community is 20% SUP05 is quite reasonable. In this scenario, sulphur oxidation accounts for the production of the majority of hydrogenases. Nevertheless, the model overestimates the abundance of sulphur oxidation genes if the SUP05 sub-population is assumed to grow by means of aerobic hydrogen oxidation (results not shown), which is discussed in detail below.
There are several possible factors that may contribute towards inappreciable growth via the Knallgas pathway. Hydrogen uptake in low concentration environments, such as at vent A1 in the ABE field, progresses at a much slower rate than in environments where H2 is abundant (Petersen et al., 2011). Therefore, spontaneous abiotic oxidation of H2—which occurs gradually over several hours—may deplete hydrogen before appreciable microbial growth via the Knallgas metabolism is able to occur (McCollom, 2000). Furthermore, H2 is also consumed as an electron donor for other metabolisms, such as sulphate reduction, methanogenesis and iron (III) reduction (McCollom, 2000). Indeed, while there are microbes that are able to grow by means of aerobic hydrogen oxidation, these organisms are typically capable of other metabolisms as well (for example, sulphur oxidation, methanotrophy; Petersen et al., 2011; Anantharaman et al., 2013). Metagenomic analysis of the microbial community in the vicinity of the ABE vent field has revealed no organism that subsists exclusively via the Knallgas metabolism (K Anantharaman, personal communication).
These simulations highlight the difficulties in inferring the occurrence of metabolisms from the presence of functional genes alone without corresponding transcriptomic or proteomic data, which could indicate which metabolisms are actually active. Nevertheless, while the Knallgas pathway may not contribute appreciably to microbial growth at vent A1, an abundance of hydrogenases mean that the community has the potential to mediate this metabolism should a hydrogen-rich environment be encountered. Metabolic versatility of this sort is likely a key ecological strategy employed by organisms that rely upon electron donors that occur in low concentrations in the deep ocean. Specifically, the propagation of metabolic genes due to growth via other pathways retains the ability to respond to different energy sources by means of a bet-hedging approach.
Impacts of hydrothermal perturbations beyond the rising plume
Electron donors that are exported from rising plumes and dispersed via physical processes represent a potential source of energy for deep-sea chemolithoautotrophs. Growth of these microbes is not only controlled by the availability of electron donors, however, but also by the energy yield of the reaction that is mediated. Multiplying the electron donor concentration at the top of the rising plume (mol l−1) by the rate of plume water export (l s−1) and energy yield per mole of electron donor (kJ mol−1) gives an estimate of energy export per unit time (kJ s−1), which is subsequently converted into an export of potential biomass via an empirical relationship (Roden and Jin, 2011), assuming these species are aerobically oxidised.
Figure 6 shows this export of potential biomass as a function of metabolic energy yield and electron donor concentration at the apex of the rising plume, assuming that all of the electron donor is used for growth. This represents an upper bound, as in nature a portion of the electron donor will be unavailable when concentrations become low. Moreover, abiotic reactions or other metabolisms not considered here may also consume the electron donor. Nevertheless, this figure allows the potential biomass production for these metabolisms to be compared relative to one another, as well as providing an upper limit on biomass. Highlighted within the plot are the metabolic pathways that are modelled above for vent A1 in the ABE field. Sulphur is shown to dominate since it is found in relatively high concentrations and is energetically profitable. In contrast, while methanotrophy yields a greater amount of energy per mole of electron donor, it is found in low concentrations meaning that it is but a minor player. Aerobic oxidation of hydrogen, ammonia and nitrite are all less energetically favourable and occur at low concentrations resulting in modest potential biomasses. Like potential biomass, the rate of growth is a function of both metabolic energy yield and enzymatic substrate availability. Consequently, metabolisms with a higher energy yield and substrate availability also grow more rapidly, which results in a temporal succession in the microbial community on the scale of weeks, as observed previously in hydrothermal plumes (Sylvan et al., 2012). When combined with transport processes, this translates into a spatial succession with maximum abundances closer to the plume source for metabolisms with higher potential biomasses, as shown below.

Export of potential biomass from the rising plume as a function of electron donor concentration and metabolic energy yield. Aerobic oxidation of ammonia, hydrogen, nitrite, methane and sulphur species for vent A1 of the ABE are plotted explicitly in the figure, assuming all of the electron donor is consumed by the considered reaction.
Data derived from drifter experiments in the Lau basin allow the various time scales of microbial growth to be cast in a spatial context. Eleven autonomous APEX and PROVOR floats were deployed in the region between April 2004 and June 2005 (Speer and Thurnherr, 2012). These autonomous drifters maintained a depth of 1700 m—comparable to the depth of neutrally-buoyant hydrothermal plumes—and surfaced every 3–4 weeks to transmit data before returning to depth again. Tracking the displacement of numerous drifters relative to their point of deployment allows probability density functions to be constructed that quantify the likelihood a water parcel is found a given distance from its source (for example, a hydrothermal plume). When divided by drift times, these distances are translated into average speeds. Finally, multiplying these speeds by a time scale of growth determines how microbial communities in the neutrally-buoyant plume have been redistributed during this period of growth. The time scale used here is doubling time as calculated by the RTM, as it provides an intuitive measure of microbial growth.
Figure 7 illustrates how microbes associated with different metabolisms have been transported away from the hydrothermal vent over their doubling time. Since sulphur oxidation occurs relatively rapidly, little dispersion has occurred by the time the associated community has doubled in size, resulting in high abundances that are concentrated close to the vent. These metabolisms also generate the greatest biomass, which contributes further to dense concentrations of sulphur oxidisers close to the vent. In stark contrast, methanotrophy is relatively slow and, therefore, methane oxidisers undergo marked dispersion while growing. In addition, methane oxidisers produce less biomass than the other metabolisms considered. Thus, while methanotroph abundances increase by an order of magnitude due to hydrothermal stimulation, slow growth rates relative to transport processes and lower biomasses relative to other microbes may mask this somewhat. Furthermore, these microbes are likely to continue grow much farther afield than sulphur oxidisers or other rapidly growing organisms.

Probability density functions showing distance of water parcels from their source (i.e., the plume) as a function of the doubling time of each metabolism.
Spatial succession due to microbial growth on different time scales is demonstrated here by tracking a water parcel along a specific trajectory. Figure 8 plots the paths of two different drifters, which were deployed close to the ABE vent field, over time and shows the predicted compositions of the microbial community at various points. Due to rapid growth and a high potential biomass, sulphur oxidisers quickly dominate the community with ammonia oxidisers accounting for the majority of other organisms present. Nevertheless, as time progresses, microbes that mediate slower metabolisms begin to appear once again, first nitrite oxidisers and then methanotrophs, albeit at modest levels due to an abundance of sulphur oxidisers persisting due to the high vent fluid concentrations and energy yields (Figure 6).

Modelled microbial communities are projected onto the trajectories (dotted lines) of two drifters released in the vicinity of the ABE vent field (denoted by a circle and cross marker). Symbols are plotted on each trajectory showing the location of the drifters at 0, 7, 168, 365 and 730 days. Corresponding to these times, pie charts show the structure of the microbial community. Background colours reflect the bathymetry of the region.
While the spatial scales shown in Figures 7 and 8 correspond to growth, microbes may endure over much larger distances depending on mortality pressures, which are notoriously difficult to estimate. The mortality rate constant used here, 0.001 d−1, was constrained for deep-sea chemolithoautotrophs by a previous study (Reed et al., 2014) and corresponds to a time scale that is much larger than those of growth (that is, 1 to nearly 3 orders of magnitude greater). These simulations point to a paradigm of microbial growth within tens of kilometers of the vent field, followed by marked persistence of microbial communities over hundreds of kilometers. As the average distance between high-temperature hydrothermal systems is <100 km (German et al., 2010), these results suggest that plumes have an important role in linking these systems. Furthermore, given the widespread distribution of hydrothermal vents along mid-ocean ridges, hydrothermal plumes potentially have a broad impact on deep-ocean biogeochemistry and microbial diversity by exchanging material with different marine systems that are hospitable to the similar microorganisms, such as oxygen minimum zones and oil spills (Dick et al., 2013). However, given that plume communities are sourced from ambient seawater and become mixed with these ambient microbial communities over time, it is difficult to delineate these two communities. Indeed, feedback between plume and ambient communities may prove an important driver of dark-ocean microbial ecology.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ . (1990). Basic Local Alignment Search Tool. J Mol Biol 215: 403–410.
Amend JP, McCollom TM, Hentscher M, Bach W . (2011). Catabolic and anabolic energy for chemolithoautotrophs in deep-sea hydrothermal systems hosted in different rock types. Geochim Cosmochim Acta 75: 5736–5748.
Anantharaman K, Breier JA, Sheik CS, Dick GJ . (2013). Evidence for hydrogen oxidation and metabolic plasticity in widespread deep-sea sulfur-oxidizing bacteria. Proc Natl Acad Sci 110: 330–335.
Anantharaman K, MB D, John A B, Kathleen A W, Brandy MT et al. (2014). Sulfur oxidation genes in diverse deep-sea viruses. Science 344: pp 757–760.
Anderson RE, Beltrán MT, Hallam SJ, Baross JA . (2012). Microbial community structure across fluid gradients in the Juan de Fuca Ridge hydrothermal system. FEMS 83: 324–339.
Arístegui J, Gasol JM, Duarte CM, Herndl GJ . (2009). Microbial oceanography of the dark ocean’s pelagic realm. Limnol Oceanogr 54: 1501–1529.
Baker BJ, Lesniewski RA, Dick GJ . (2012). Genome-enabled transcriptomics reveals archaeal populations that drive nitrification in a deep-sea hydrothermal plume. ISME J 6: 2269–2279.
Baker BJ, Sheik CS, Taylor CA, Jain S, Bhasi A, James D C et al. (2013). Community transcriptomic assembly reveals microbes that contribute to deep-sea carbon and nitrogen cycling. ISME J 7: 1962–1973.
Boettger Jason, Lin Huei Ting, Cowen James P, Hentscher Michael, Amend Jan P . (2013). Energy yields from chemolithotrophic metabolisms in igneous basement of the Juan de Fuca ridge flank system. Chem Geol 337–338: 11–19.
Breier JA, Rauch CG, McCartney K, Toner BM, Fakra SC, White SN et al. (2009). A suspended-particle rosette multi-sampler for discrete biogeochemical sampling in low-particle-density waters. Deep Sea Res 56: 1579–1589.
Breier JA, Toner BM, Fakra SC, Marcus MA, White SN, Thurnherr AM et al. (2012). Sulfur, sulfides, oxides and organic matter aggregated in submarine hydrothermal plumes at 9°50’N East Pacific Rise. Geochim Cosmochim Acta 88: 216–236.
Breier JA, Sheik CS, Gomez-Ibanez D, Sayre-McCord RT, Sanger R, Rauch C et al. (2014). A large volume particulate and water multi-sampler with in situ preservation for microbial and biogeochemical studies. Deep Sea Res I 94: 195–206.
Converse DR, Holland HD, Edmond JM . (1984). Flow rates in the axial hot springs of the East Pacific Rise (21° N) implications for the heat budget and the formation of massive sulfide deposits. Earth Planet Sci Lett 69: 159–175.
Dick Gregory J, Anantharaman Karthik, Baker Brett J, Li Meng, Reed Daniel C, Sheik Cody S . (2013). The microbiology of deep-sea hydrothermal vent plumes: ecological and biogeographic linkages to seafloor and water column habitats. Front Microbiol 4: 124.
Dick Gregory J, Tebo Bradley M . (2010). Microbial diversity and biogeochemistry of the Guaymas Basin deep-sea hydrothermal plume. Environ Microbiol 12: 5.
Dick Jeffrey M . (2008). Calculation of the relative metastabilities of proteins using the CHNOSZ software package. Geochemical Transactions 9.10. Available from: http://www.geochemicaltransactions.com/content/9/1/10.
Dick Jeffrey M, Shock Everett L . (2013). A metastable equilibrium model for the relative abundances of microbial phyla in a hot spring. PLoS One 8: 9.
Falkowski Paul G, Fenchel Tom, Delong Edward F . (2008). The microbial engines that drive earth’s biogeochemical cycles. Science 320: 1034–1039.
Flores GE, Shakya M, Meneghin J, Yang ZK, Seewald JS, Wheat CG et al. (2012). Inter-field variability in the microbial communities of hydrothermal vent deposits from a back-arc basin. Geobiology 10: 333–346.
German CR, Thurnherr AM, Knoery J, Charlou J.-L, Jean-Baptiste P, Edmonds HN . (2010). Heat, volume and chemical fluxes from submarine venting: a synthesis of results from the Rainbow hydrothermal field, 36°N MAR. Deep Sea Res 57: 518–527.
Gilbert Jack A, Dupont Christopher L . (2011). Microbial metagenomics: beyond the genome. Annu Rev Mar Sci 3: 347–371.
Grassle J. Fredrick . (1986). The ecology of deep-sea hydrothermal vent communities. Adv Mar Biol 23: 301–362.
Helfrich KR, Speer KG . (1995). Oceanic hydrothermal circulation: mesoscale and basin-scale flow. Seafloor hydrothermal systems: physical, chemical, biological, and geological interactions In: Humphris SE, Zierenberg RA, Mullineaux LS, Thomson RE (eds). American Geophysical Union: Washington, DC; doi:10.1029/GM091p0347.
Huber Julie A, Butterfield David A, Baross John A . (2003). Bacterial diversity in a subseafloor habitat following a deep-sea volcanic eruption. FEMS 43: 393–409.
IPCC. (2013). In Stocker TF, Qin D, Plattner G-K, Tignor M, Allen SK, Boschung J et al. eds. Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovern- mental Panel on Climate Change. Cambridge University Press: Cambridge, UK; New York, NY, USA, p 1535.
Jannasch Holger W, Wirsen Carl O . (1979). Chemosynthetic primary production at east Pacific Sea floor spreading centers. BioScience 29: 592–598.
Jiang Houshuo, Breier John A . (2014). Physical controls on mixing and transport within rising submarine hydrothermal plumes: a numerical simulation study. Deep Sea Res I 92: 41–55.
Jin Qusheng, Bethke Craig M . (2005). Predicting the rate of microbial respiration in geochemical environments. Geochim Cosmochim Acta 69: 1133–1143.
Karner Markus B, DeLong Edward F, Karl David M . (2001). Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 409: 507–510.
LaRowe Douglas E, Dale Andrew W, Aguilera David R, Ivan L’Heureux Jan P, Amend, Regnier Pierre . (2014). Modeling microbial reaction rates in a submarine hydrothermal vent chimney wall. Geochim Cosmochim Acta 124: 72–97.
Lesniewski Ryan A, Jain Sunit, Anantharaman Karthik, Schloss Patrick D, Dick Gregory J . (2012). The metatranscriptome of a deep-sea hydrothermal plume is dominated by water column methanotrophs and lithotrophs. ISME J 6: 2257–2268.
Li M, Toner BM, Baker BJ, Breier JA, Sheik CS, Dick GJ . (2014). Microbial iron uptake as a mechanism for dispersing iron from deep-sea hydrothermal vents. Nat Commun 5: 3192.
Lupton JE, Delaney JR, Johnson HP, Tivey MK . (1985). Entrainment and vertical transport of deep- ocean water by buoyant hydrothermal plumes. Nature 316: 621–623.
Marshall KT, Morris RM . (2013). Isolation of an aerobic sulfur oxidizer from the SUP05/Arctic96BD-19 clade. ISME J 7: 452–455.
Martens-Habbena Willm, Berube Paul M, Urakawa Hidetoshi, de la Torre José R, Stahl David A . (2009). Ammonia oxidation kinetics determine niche separation of nitrifying Archaea and Bacteria. Nature 461: 976–981.
McCollom Thomas M . (2000). Geochemical constraints on primary productivity in submarine hydrothermal vent plumes. Deep Sea Res 47: 85–101.
McCollom Thomas M . (2007). Geochemical constraints on sources of metabolic energy for chemolithoautotrophy in ultramafic-hosted deep-sea hydrothermal systems. Astrobiology 7: 933–950.
McCollom Thomas M, Shock Everett L . (1997). Geochemical constraints on chemolithoautotrophic metabolism by microorganisms in seafloor hydrothermal systems. Geochim Cosmochim Acta 61: 4375–4391.
McDuff RE . (1995). Physical dynamics of deep-sea hydrothermal plumes, in seafloor hydrothermal systems: physical, chemical, biological, and geological interactions. In: Humphris SE, Zierenberg RA, Mullineaux LS, Thomson RE (eds). American Geophysical Union: Washington, DC; doi:10.1029/GM091p0357.
Mottl Michael J, Seewald Jeffrey S, Geoffrey Wheat C, Margaret K Tivey, Peter J Michael, Proskurowski Giora et al. (2011). Chemistry of hot springs along the Eastern Lau Spreading Center. Geochim Cosmochim Acta 75: 1013–1038.
Nakamura Kentaro, Takai Ken . (2014). Theoretical constraints of physical and chemical properties of hydrothermal fluids on variations in chemolithotrophic microbial communities in seafloor hydrothermal systems. Prog Earth Planet Sci 1: 5.
Nogales B, Lanfranconi MP, Piña-Villalonga JM, Bosch . (2013). Anthropogenic perturbations in marine microbial communities. FEMS Microbiol Rev 35: 275–298.
Petersen Jillian M, Zielinski Frank U, Pape Thomas, Seifert Richard, Moraru Cristina, Amann Rudolf et al. Hydrogen is an energy source for hydrothermal vent symbioses. (2011) Nature 476: 176–180.
R Development Core Team. (2006). R: A Language and Environment for Statistical Computing. ISBN 3-900051-07-0. R Foundation for Statistical Computing. Vienna, Austria. Available from: http://www.R-project.org.
Reed Daniel C, Algar Christopher K, Huber Julie A, Dick Gregory J . (2014). Gene-centric approach to integrating environmental genomics and biogeochemical models. PNAS 111: 1879–1884.
Roden Eric E, Jin Qusheng . (2011). Thermodynamics of microbial growth coupled to metabolism of glucose, ethanol, short-chain organic acids, and hydrogen. Appl Environ Microbiol 77: 1907–1909.
Sheik Cody S, Anantharaman Karthik, Breier John A, Jason B Sylvan, Gregory J Dick Deep ocean background microbial communities are primary colonizers of hydrothermal plumes across a back-arc spreading basin. ISME J (in press); doi:10.1029/GM091p0347.
Shock Everett L, McCollom Thomas M, Schulte Micthell D . (1995). Geochemical constraints on chemolithoautotrophic reactions in seafloor hydrothermal systems. Origins Life Evol B 25: 141–159.
Soetaert Karline, Meysman Filip . (2012). Reactive transport in aquatic ecosystems: rapid model prototyping in the open source software R. Environ Model Softw 32: 49–60.
Soetaert Karline, Thomas Petzoldt R, Woodrow Setzer . (2010). Solving differential equations in R: Package deSolve. J Stat Softw 33.9 pp, 1–25 ISSN: 1548-7660. Available from: http://www.jstatsoft.org/v33/i09.
Speer Kevin, Thurnherr Andreas . (2012). The Lau Basin Float Experiment (LAUB-FLEX). Oceanography 25: 284–285.
Stolper Daniel A, Revsbech Niels Peter, Canfield Donald E . (2010). Aerobic growth at nanomolar oxygen concentrations. Proc Natl Acad Sci 107: 18755–18760.
Sunamura Michinari, Higashi Yowsuke, Miyako Chiwaka, Ishibashi Jun-ichiro, Maruyama Akihiko . (2004). Two bacteria phylotypes are predominant in the Suiyo Seamount Hydrothermal Plume. Appl Environ Microbiol 70: 1190–1198.
Sylvan JB, Pyenson BC, Rouxel O, German CR, Edwards KJ . (2012). Time-series analysis of two hydrothermal plumes at 9°50’N East Pacific Rise reveals distinct, heterogeneous bacterial populations. Geobiology 10: 178–192.
Tagliabue Alessandro, Bopp Laurent, Dutay Jean-Claude, Bowie Andrew R, Chever Fanny, Philippe Jean-Baptiste et al. (2010). Hydrothermal contribution to the oceanic dissolved iron inventory. Nat Geosci 3: 252–256.
Tivey Margaret Kingston . (2004) Environmental Conditions Within Active Seafloor Vent Structures: Sensitivity to Vent Fluid Composition and Fluid Flow. Vol. Geophysical Monograph Series 144. The Sub-seafloor Biosphere at Mid-Ocean Ridges. American Geophysical Union: Washington DC, pp 137–152.
Winn Christopher D, Karl David M, Massoth Gary J . (1986). Microorganisms in deep-sea hydrothermal plumes. Nature 320: 744–746.
Acknowledgements
This work is funded by Gordon and Betty Moore Foundation grant GBMF2609 and NSF grant OCE 1038006 to GJD, NSF grants OCE-0241785 and OCE-1231803 to KS, NSF grant OCE-1037991 to BMT, and OCE-1038055 to JAB and HJ. We thank the Captain and crew of the R/V Thomas G Thompson as well as the crew of the ROV Jason II. Thanks to Drs AL Reysenbach, M Tivey, C Fisher, P Girguis, G Luther and the Eastern Lau Spreading Center 2009 scientific parties for allowing us to participate in their cruises (NSF grants: OCE-0424953, OCE-02040985, OCE-0728391, OCE-0752469 and OCE-0751839). Also, we thank Drs Sheri White and Jason Sylvan for assistance with sampling. Nucleotide sequences are available in the NCBI Sequence Read Archive, accession numbers SRX511269, SRX511304 and SRX511275 (Supplementary Table S4). These data have been previously published by Anantharaman et al. (2014). The custom databases, blastx results and reads that hit each gene referenced in this study are available at the following URL along with the model code: www.earth.lsa.umich.edu/geomicrobiology/Lau.html
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interests.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Reed, D., Breier, J., Jiang, H. et al. Predicting the response of the deep-ocean microbiome to geochemical perturbations by hydrothermal vents. ISME J 9, 1857–1869 (2015). https://doi.org/10.1038/ismej.2015.4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.4
This article is cited by
-
Strategies of chemolithoautotrophs adapting to high temperature and extremely acidic conditions in a shallow hydrothermal ecosystem
Microbiome (2023)
-
Sulfur cycling connects microbiomes and biogeochemistry in deep-sea hydrothermal plumes
The ISME Journal (2023)
-
Niche differentiation of sulfur-oxidizing bacteria (SUP05) in submarine hydrothermal plumes
The ISME Journal (2022)
-
Hydrothermal plumes as hotspots for deep-ocean heterotrophic microbial biomass production
Nature Communications (2021)
-
Microbial invasions in sludge anaerobic digesters
Applied Microbiology and Biotechnology (2021)
