
Abstract
Reef-building corals form complex relationships with a range of microorganisms including bacteria, archaea, fungi and the unicellular microalgae of the genus Symbiodinium, which together form the coral holobiont. These symbionts are known to have both beneficial and deleterious effects on their coral host, but little is known about what the governing factors of these relationships are, or the interactions that exist between the different members of the holobiont and their environment. Here we used 16S ribosomal RNA gene amplicon sequencing to investigate how archaeal and bacterial communities associated with the widespread scleractinian coral Seriatopora hystrix are influenced by extrinsic (reef habitat and geographic location) and intrinsic (host genotype and Symbiodinium subclade) factors. Bacteria dominate the microbiome of S. hystrix, with members of the Alphaproteobacteria, Gammaproteobacteria and Bacteriodetes being the most predominant in all samples. The richness and evenness of these communities varied between reef habitats, but there was no significant difference between distinct coral host lineages or corals hosting distinct Symbiodinium subclades. The coral microbiomes correlated to reef habitat (depth) and geographic location, with a negative correlation between Alpha- and Gammaproteobacteria, driven by the key members of both groups (Rhodobacteraceae and Hahellaceae, respectively), which showed significant differences between location and depth. This study suggests that the control of microbial communities associated with the scleractinian coral S. hystrix is driven primarily by external environmental conditions rather than by those directly associated with the coral holobiont.
Similar content being viewed by others


Introduction
Reef-building corals form complex relationships with a range of different microbial partners, including bacteria, archaea and algae belonging to the genus Symbiodinium, which together form the coral holobiont. The genus Symbiodinium can be categorised into eight distinct phylogenetic clades (A–H) based on their ribosomal DNA sequence (ITS2), with five of these (A–D and F) found in association with the corals (Baker, 2003). A high degree of host specificity (LaJeunesse et al., 2004, 2010), habitat partitioning and ecological diversification have been identified as controlling factors of the distribution of the different Symbiodinium clades (Douglas, 1998; LaJeunesse et al., 2003; LaJeunesse, 2005; Stat et al., 2008a; Bongaerts et al., 2010, 2011b; LaJeunesse et al., 2010; Silverstein et al., 2011). To date the main role of Symbiodinium in the coral holobiont is thought to be through its involvement in its host’s metabolism, including photosynthesis and nutrient cycling, which provides their hosts with up to 95% of their carbon requirements (Muscatine and Porter, 1977) and the cycling of nitrogenous compounds (Leggat et al., 2007; Pernice et al., 2012).
Coral-associated bacteria and archaea have been extensively studied for over four decades with thousands of species having been identified (Mouchka et al., 2010). However, we are only just starting to investigate and understand the forces that govern their patterns of diversity, distribution and role in the coral holobiont ecosystem. Bacteria, archaea and viruses are ubiquitous in the marine system, and thrive in coral reef waters (Sorokin, 1973; Dinsdale et al., 2008), but these communities have been shown to be distinct from those associated with the coral animal (Rohwer et al., 2001, 2002; Cooney et al., 2002; Frias-Lopez et al., 2002; Guppy and Bythell, 2006; Santiago-Vazquez et al., 2007; Kvennefors et al., 2010; Sunagawa et al., 2010; Chen et al., 2011; Tremblay et al., 2011; Ceh et al., 2012; Schöttner et al., 2012), suggesting that there is specific control of the microbiomes (associated bacterial and archaeal communities), by either biological or microenvironmental factors influenced by the host.
The complex physical colony structure of the coral host provides a multi-faceted habitat with different microbial communities occupying a range of niches within the surface mucus layer, tissue layers (Paul et al., 1986; Ritchie and Smith, 1995b; Banin et al., 2000; Johnston and Rohwer, 2007; Lesser et al., 2007; Sharp et al., 2010, 2012) and in the different zones of the colony (Rohwer et al., 2002; Bourne and Munn, 2005). As with terrestrial ecosystems (Balser et al., 2006), coral reef-associated microbes are thought to drive biochemical and ecological processes including nitrogen (Williams et al., 1987; Shashar et al., 1994; Ritchie and Smith, 1995b; Lesser et al., 2004; Lesser et al., 2007; Olson et al., 2009; Kimes et al., 2010; Lema et al., 2012) and carbon cycling (Ducklow and Mitchell, 1979; Ritchie and Smith, 1995b; Wild et al., 2004b, 2010b), allowing reefs to prosper in the oligotrophic waters of the tropics. In addition to their role in driving ecosystem function through nutrient cycling, microbes also have an important role in coral health. For example, they are able to contribute to pathogen inhibition through competition for space and nutrients, and the secretion of antibiotic substance (Koh, 1997; Castillo et al., 2001; Ritchie, 2006; Rypien et al., 2010; Sharp et al., 2010; Kvennefors et al., 2012), as well as potentially aiding reproduction and propagation (Apprill et al., 2009; Sharp et al., 2010, 2012). Less is known about the role of archaea associated with reef-building corals. Archaea associated with the coral tissues have been found to occur at varying cell densities (Wegley et al., 2004), and with varied levels of diversity relative to bacterial communities (Wegley et al., 2004, 2007; Siboni et al., 2008; Lins-de-Barros et al., 2010; Littman et al., 2011), and in some cases have been found to be absent from coral colony tissues altogether (Yakimov et al., 2006; Hansson et al., 2009). Coral-associated archaeal communities are also independent of those in the water column, but in contrast to bacteria they are fairly cosmopolitan, displaying no evidence of species specificity. Despite their apparent generality it is thought that archaea still have an important role in the life of their host through nutrient cycling (Wegley et al., 2004; Beman et al., 2007; Siboni et al., 2008, 2012).
Evidence to date suggests that the control of coral-associated bacterial and archaeal communities may be a result of multiple factors and at different spatial scales, however, not all studies are in agreement as to the primary controlling factor. Environmental factors such as temperature and nutrients, which may differ between location and season, have been found to significantly influence the specificity of bacterial–coral associations (Guppy and Bythell, 2006; Klaus et al., 2007; Hong et al., 2009; Littman et al., 2009b; Schöttner et al., 2012). In addition, species-specific control where bacterial communities were specifically associated with corals of the same species across distinct geographical regions at different times has been seen (Frias-Lopez et al., 2002; Rohwer et al., 2002; Kvennefors et al., 2010). It is thought that the control of associated microbial communities at this level is due to the differences in coral species-specific mucus properties (Ducklow and Mitchell, 1979; Meikle, et al., 1988; Ritchie and Smith, 2004; Wild et al., 2004a, 2010a; Ritchie, 2006; Tremblay et al., 2011; Schöttner et al., 2012), which in turn is strongly linked to physiological differences between coral species. Nonetheless, it remains unclear whether the specificity of microbial communities extends to the host intraspecific level (differences in the microbiomes of closely related coral lineages) and whether the observed microbiome differences are influenced by the type of Symbiodinium present in the coral (which also contributes substantially to the metabolic makeup of the mucus (Kellogg and Patton, 1983; Crossland, 1987; Anthony and Fabricius, 2000).
Here, the microbiomes of the scleractinian coral Seriatopora hystrix were assessed in a multi-factorial design, testing for differences related to coral host genotype (based on the putative control region of the coral mitochondrial DNA), Symbiodinium subclade genotype (based on the ITS2 region of the algal ribosomal DNA), reef habitat (depth) and geographic location. Samples were collected from three habitats (‘Back Reef’, ‘Upper Slope’ and ‘Deep Slope’) and at three locations on the Northern Great Barrier Reef (Yonge Reef, Day Reef and Lizard Island). Host-symbiont assemblages were genotyped in a previous study (Bongaerts et al., 2010), representing a total of five distinct host–Symbiodinium combinations that allowed the assessment of the individual effects of host and Symbiodinium genotype (one host genotype associated with three different Symbiodinium subclades and one Symbiodinium subclade found across two different host genotypes). Similarly, although these host–Symbiodinium genotypes were partitioned across habitats, genotype–environment ‘mismatches’ (for example, a ‘shallow’ genotype occurring in the ‘Deep Slope’ habitat) allowed us to assess the individual effects of host–Symbiodinium genotype and environment. Microbial communities were characterised using 16S ribosomal RNA gene amplicon sequencing, and the individual contributions to richness and evenness were assessed across distinct host lineages, symbiont genotypes, reef habitats and geographic locations.
Materials and methods
Sample collection and surveys
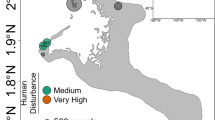
Corals were identified as Seriatopora hystrix based on characters described by Veron (2000) and Veron and Pichot (1976). Small fragments of Seriatopora colonies were collected as described in Bongaerts et al. (2010) from three different habitats (Figure 1): the ‘Back Reef’ (2 m depth±1 m), ‘Upper Slope’ (6 m depth±1 m) and ‘Deep Slope’ (27 m depth±2 m) at two reef locations, Yonge Reef (14°36′59.9″S; 145°38′11.1″E) and Day Reef (14°28′28.4″S;145°32′19.1″E) along the continental shelf edge of the Great Barrier Reef, and from a ‘Back Reef’ habitat (2 m±1 m) at Lizard Island (14°41′39.1″S; 145°27′58.2″E). All sampled colonies were separated by at least 3 m in order to minimise the inclusion of potential clone mates due to fragmentation. Coral tissue was separated from the coral skeleton with a modified air gun attached to a SCUBA cylinder and subsequently stored in 20% dimethyl sulfoxide preservation buffer and kept at −20 °C until further processing.

Study locations, habitats and Seriatopora hystrix coral–Symbiodinium assemblages. (a) Studied coral host–Symbiodinium assemblages across the three different habitat types: ‘Back Reef’, ‘Upper Slope’ and ‘Deep Slope’. Inset: map of the three study locations on the northern Great Barrier Reef: Lizard Island, Yonge Reef and Day Reef. (b) Diversity and distribution of host and Symbiodinium genotypes across the three habitats and locations. Symbol shape refers to host genotype, and colour refers to symbiont genotype. Asterisk indicates assemblages not included in this study.
Photographs were taken of random colonies in the three depth habitats at the time of sampling at Yonge Reef in 2008 (n=40) and again during the ‘Catlin Seaview Survey’ in 2012 when both Yonge and Day Reef were revisited (n=44 and n=45, respectively), and visually assessed for level of pigmentation (‘pigmented’ vs ‘pale’).
Host–Symbiodinium genotyping and sample selection
Genotyping of the corals and associated Symbiodinium was performed on total genomic DNA extracts as described in Bongaerts et al. (2010). Here, we selected 11 different combinations of host genotype, Symbiodinium genotype, reef habitat (depth) and location for which triplicate samples (each from a different coral colony) were available (Figure 1b). Five different host–Symbiodinium genotype combinations were assessed, representing three different host genotypes (mtDNA genotypes: B, U and D1) and four different Symbiodinium ITS2 subclades (C120, C120a, C1m-aa and C3n-t; Figure 1b). Although combinations were partitioned across habitats, there was some overlap in genotypes (that is, HostU:C120 occurring in the ‘Upper Slope’ and ‘Deep Slope’ habitats) which allowed us to test for the effect of habitat independent of genotype. Similarly, in the ‘Back Reef’ habitat a single host genotype with three different Symbiodinium types were found at Day Reef, allowing us to test for the independent effect of Symbiodinium type.
Analysis of bacterial and archaeal community composition by amplicon sequencing
Primers broadly targeting all bacteria and archaea containing the Roche 454 adaptor ligated at the 5′ end were used to amplify the V6–V8 region of the 16S ribosomal RNA gene: pyroLSSU803F 5′-TTAGAKACCCBNGTAGTC-3′ and pyroLSSU1392R 5′-ACGGGCGGTGWGTRC-3′. Amplification reaction mixture (50 μl) included: × 10 PCR buffer (5 μl); 1 μl dNTPs (10 mM each); 4 μl MgCl (25 mM); 1 μl each primer (10 μM); 0.2 μl Taq DNA Polymerase (Thermo Fisher Scientific, Scoresby, VIC, Australia); 1.5 μl bovine serum albumin; ∼20 ng DNA template; and made up to 50 μl with RNA/DNA-free water. Reaction conditions were as follows: an initial denaturation was carried out at 95 °C for 3 min followed by 30 cycles of denaturation at 95 °C for 30 sec; annealing at 55 °C for 30 sec; and primer extension at 74 °C for 30 sec. This was followed by a final extension step at 74 °C for 10 min. Amplicon size section was carried out using Pippin Prep System (Sage Science, Beverly, MA, USA) before sequencing on a Roche 454 GS FLX (Roche, Basel, Switzerland) at the Australian Centre for Ecogenomics, University of Queensland, Australia. The sequence data set was deposited in the NCBI Sequence Read Archive (SRA) database (accession numbers: SRR1664591–SRR1664608, SRR1664610–SRR1664614 and SRR1664616–SRR1664625).
Sequence data processing and statistical analysis
Sequences were quality filtered and dereplicated with the QIIME script split_libraries.py with the homopolymer filter deactivated (Caporaso et al., 2010) and then checked for chimeras against the 2012 edition of the Greengenes database (DeSantis et al., 2006) with UCHIME ver. 3.0.617 (Edgar et al., 2011) as previously described (Dennis et al., 2013). Homopolymer errors were corrected with Acacia (Bragg et al., 2012). Sequences were then subjected to the following procedures with QIIME scripts at the default settings. (i) Sequences were clustered at 97% similarity using UCLUST ver. 3.0.617 (Edgar et al., 2011, ii) cluster representatives were selected, (iii) greengenes taxonomy (2012 edition) was assigned to the cluster representatives by BLAST and (iv) tables with the abundance of different operational taxonomic units (OTUs) and their taxonomic assignments in each sample were generated. All reads matching eukaryotes or chloroplasts were removed and the number of reads was normalised to 1900 per sample. The mean number of OTUs (observed richness) and Simpson diversity index values (Simpson, 1949) corresponding to 1900 sequences per sample were calculated with QIIME. Generalised linear modelling was used to assess whether variation in observed richness and Simpson diversity index values could be explained by any of our treatments (that is, reef, depth, host genotype and Symbiodinium subclade). Differences in the composition of microbial communities between samples were assessed using permutational multivariate analysis of variance (PERMANOVA). Richness and evenness were compared using Tukey’s Honestly Significant Difference (HSD) tests. All analyses were implemented with R version 2.12.0 (R Foundation for Statistical Computing, Vienna, Austria). Sequences were aligned against the greenegenes nucleotide database (version September 2014) in ARB using PyNAST (Ludwig et al., 2004). Neighbour-joining maximum likelihood phylogenetic trees were constructed in FastTree v2.1.7 (Lawrence Berkeley National Lab, Berkeley, CA, USA).
Results and Discussion
Three representative samples (one per colony) of each combination at each site were amplified and sequenced, producing an average of 1900 reads per sample. The alpha diversity of the microbial communities as described by the Simpson Index and OTU richness associated with the Seriatopora hystrix colonies demonstrated a wide range of variation (Figure 2 and Supplementary Table S1) with the Simpson’s index ranging from 0.265 to 0.988, and up to 705 OTUs. Archaeal populations comprised <1.5% relative abundance of coral microbiomes in all samples, with most OTUs belonging to the Crenarchaeota. The bacterial communities were dominated by members of the Alpha- and Gammaproteobacteria at all locations and depths as reported in other studies (Klaus et al., 2007; Littman et al., 2009a, 2009b; Olson et al., 2009; Kvennefors et al., 2012). In particular, members of the alphaproteobacterial family Rhodobacteraceae and gammaproteobacterial genus Endozoicomonas were the most abundant OTUs. Phylogenetic analysis showed that the most abundant Endozoicomonas OTU clustered (Figure 3a) with sequences from cultivated and uncultivated species previously found in eight other scleractinian corals (Kvennefors et al., 2010; Sunagawa et al., 2010; Yang et al., 2010; Speck and Donachie, 2012; Apprill et al., 2013; Bayer et al., 2013; Jessen et al., 2013), as well as soft corals, sea slugs, sea anemones and sea cucumbers (Kurahashi and Yokota, 2007; Schuett et al., 2007). Bayer et al. (2013) identified Endozoicomonas cells in high densities within the tissues of the coral Stylophora pistillata where it formed aggregations in close proximity to the endosymbiotic Symbiodinium in the endoderm, resembling the ovoid bacterial clusters found in other studies (Peters, 1984; Santavy and Peters, 1997; Ainsworth and Hoegh-Guldberg, 2009). Due to their high densities and intimate relationship with the coral tissues and algal endosymbionts it is thought that they may provide an advantage to their host’s health through the production of antimicrobial compounds (Ritchie, 2006). Principal component analysis identified 17 OTUs, in addition to the Endozoicomonas OTUs (OTU 69 and 71), which were responsible for driving the differences in microbial communities associated with the different samples. These other OTUs were related to members of the Rhodobacterales and Vibrionales, several members of which have previously been identified as putative pathogens, or associated with disease lesions of scleractinian corals and other marine invertebrates (Imai et al., 2006; Koren and Rosenberg, 2008; Becker et al., 2009; Sekar et al., 2009; Sunagawa et al., 2009; Case et al., 2011; Fernandes et al., 2011; Figure 3b). OTUs from these groups were also found to have a highly significant negative correlation to Endozoicomonas. For example, the Endozoicomonas OTU 69 demonstrated a very strong negative correlation to OTU 25 (r=−0.73, P<0.01), with moderate-to-strong negative correlations to OTUs 51, 30, 28 and 21 (r=−0.58, −0.39, −0.61 and −0.5 respectively, P<0.05).

Heatmap summarising the alpha diversity and population abundances of dominant bacterial and archaeal OTUs (those present at >1% relative abundance) associated with Seriatopora hystrix colonies from Day Reef, Yonge Reef and Lizard Island Reef with different host–Symbiodinium genotype combinations. A total of three samples per location habitat and host–Symdiodinium genotype combination were analysed. Symbol shape refers to host genotype, and colour refers to symbiont genotype.

Phylogenetic trees, based on 16S ribosomal RNA gene sequences, showing the positions of OTUs identified associated with Seriatopora hystrix colonies relative to previously identified ribotypes obtained from public databases. (a) Position of OTUs 69 and 71 relative to members of the genus Endozoicomonas identified associated with other marine organisms including six Scleractinian corals. Scale bar represents 0.1 changes per nucleotide. (b) Position of OTUs 25, 28, 30, 51 and 76 relative to previously identified marine pathogens. Scale bar represents 0.1 changes per nucleotide. GenBank accession numbers are shown in parentheses. Numbers at nodes are bootstrap percentages (of 100 resampled data sets); only ⩾75% are shown.
Effect of eukaryotic holobiont members on associated microbial communities
Microbes are highly sensitive to their chemical microenvironment. Due to this and their intimate relationship with the coral tissues it was expected that community structure would change with host and Symbiodinium genotype and the associated physiological differences as seen in previous studies (Ikeda and Miyachi, 1995; Ritchie and Smith, 1995a, 1995b; Ritchie, 2006; Littman et al., 2009a; Raina et al., 2010; Tremblay et al., 2011). However, neither host nor Symbiodinium differences were found to influence the associated microbial communities. The microbial communities associated with the dominant host–Symbiodinium assemblages of ‘Upper Slope’ and ‘Deep Slope’ habitats (HostU:C120 and HostD1:C3n-t, respectively) were not found to be significantly different when co-occurring at the same depth (that is, HostU:C120 occurs in low abundances in the ‘Deep Slope’ habitat). This, despite the strong physiological differences observed for these host–Symbiodinium assemblages, even when growing in the same habitat (Bongaerts et al., 2011a). The ‘Back Reef’ coral communities of Day Reef were made up of three different host–Symbiodinium genotype assemblages, consisting of the same host genotype (HostU) and three independent algal subclades (C120, C120a and C1m-aa). Comparison of the three assemblage types demonstrated also no difference in community structure with Symbiodinium genotype. The close genetic relationship of the three subclades present within the ‘Back Reef’ corals at Lizard Island (Bongaerts et al., 2011a) may mean that they have similar physiologies and consequently do not produce significantly different photosynthetic products, although identification of differences in photosynthate composition at the subclade level remains to be carried out.
Effect of reef location on coral-associated microbial communities
Ribbon reefs at the edge of the continental shelf such as Yonge and Day Reef experience substantially different environment conditions (for example, temperature and nutrients) to mid-shelf reefs like Lizard Island due to the direct exposure to deep oceanic waters (Berkelmans et al., 2004; De'ath and Fabricius, 2010; Fabricius et al., 2014). These environmental differences may have a strong influence on the coral-associated microbial communities at each location. The microbial communities associated with S. hystrix colonies from the ‘Back Reef’ habitat (HostB:C120) were significantly different at the three locations (PERMANOVA, F2,6=2.48, P<0.005). Lizard Island ‘Back Reef’ communities were dominated by members of the Alphaproteobacteria (OTUs 24 and 25, Figure 2), while Yonge Reef ‘Back Reef’ communities had a greater relative abundance of members of the gammaproteobacterial genus Endozoicomonas (OTU 69, Figure 2). The microbial communities associated with the ‘Back Reef’ colonies at Day Reef shared similarities with both Yonge and Lizard Island Reefs with higher relative abundance of members of both the Rhodobacteraceae and Endozoicomonas as well as a high relative abundance of other Alphaproteobacteria. Significant differences also existed in the alpha diversity between the different ‘Back Reef’ habitats (one-way analysis of variance: Sobs: F2,6=9.022, P<0.05; Simpson Diversity: F2,6=9.022, P<0.01). The richness and evenness (Supplementary Table S1 and Supplementary Figure S1) of microbial communities from the ‘Back Reef’ of Lizard Island reef were significantly lower than those of Day Reef (Tukey’s HSD post hoc: Sobs P<0.05; Simpsons Diversity: P<0.01), and the evenness was significantly lower to those of Yonge Reef (Tukey’s HSD post hoc: Simpson’s diversity: P<0.05). These differences may be due to varying levels of habitat heterogeneity of the mid- and outer-shelf reefs. Heterogeneity has been shown to be one of the main determinants of community structure (Jiang and Patel, 2008). It may therefore be expected that the higher levels of disturbance and extremes of conditions such as temperature and solar irradiance (Bongaerts et al., 2011a) experienced in ‘Back Reef’ environments may lead to specialisation of communities and reduced diversity (Buckling et al., 2000; Berga et al., 2012). Day and Yonge Reefs however may not be exposed to the same levels of extremes as Lizard Island due to their proximity to the shelf edge and exposure to oceanic waters, which is also reflected in the differential bleaching susceptibility of reefs depending on the shelf position (Berkelmans and Oliver, 1999).
As no effect of host-Symbiodinium genotype was found within ‘Deep Slope’ habitats (that is, between HostU:C120 and HostD1:C3n-t genotypes), samples from these habitats were grouped and compared between locations (that is, Day and Yonge Reef). A significant difference (PERMANOVA, F1,9=2.58, P<0.05) was found between ‘Deep Slope’ coral microbiomes from Yonge Reef and Day Reef, but no difference was found in their alpha diversity. ‘Deep Slope’ communities were dominated by Alphaproteobacteria with differences driven by a greater prevalence of members of the Gammaproteobacteria, with Vibrionaceae and Alteromonadaceae associated with the Day Reef corals than those from Yonge Reef, which had a greater occurrence of Pseudoalteromonadaceae (Figure 2). In contrast, for the ‘Upper Slope’ habitat (HostU:C120 genotype), no significant difference was found in the microbial communities of Day and Yonge Reef, which were dominated by Endozoicomonas (Figure 2, OTU 69).
Previous studies have also found that geographical differences exist between the microbial communities associated with corals (Rohwer et al., 2002; Guppy and Bythell, 2006; Klaus et al., 2007; Littman et al., 2009b; Chen et al., 2011; Schöttner et al., 2012). The potential pathogen-dominated communities associated with ‘Back Reef’ colonies at Lizard Island may be due to their closer proximity to human influences (Dinsdale et al., 2008; Furby et al., 2014) and reduced influence of oceanic waters due to their position in the mid-shelf. Although Yonge and Day Reef ‘Deep Slope’ coral microbiomes possessed the same group of Rhodobacteraceae related to potential pathogens, those of the Day Reef had a greater abundance of Endozoicomonas (Figure 2, OTU 69). At the time the samples were collected (October 2008) it was found that in contrast to the shallow habitats, deep reef habitats (∼27 m depth) experienced a more constant environmental state, in terms of more consistent and lower levels of irradiance due to light attenuation through the water column and reduced thermal variability compared with the diurnal fluctuations experienced in the ‘Back Reef’ habitats (although in summer, the ‘Deep Reef’ habitat can experience cold-water influxes; Bongaerts et al., 2011a). It would also be expected that the Deep Reef sites would also experience reduced wave stress in comparison with the shallow habitats. These more homogeneous and stable conditions may allow location-specific factors not measured as a part of this study to have a role in shaping the microbial communities, resulting in differences between communities associated with deep-water S. hystrix colonies.
Effect of depth habitat on associated microbial communities
S. hystrix colonies with the HostU:C120 genotype can be found at both the ‘Upper Slope’ and ‘Deep Slope’ habitats (albeit in low abundance in the latter) of Day and Yonge Reef, and their microbial communities were found to be significantly different between depths (PERMANOVA, F1,9=4.398, P<0.001). The community alpha diversity also differed significantly with depth, with ‘Deep Slope’ S. hystrix microbiomes being considerably more diverse and even than those of the ‘Upper Slope’ colonies (one-way analysis of variance: richness F2,9=5.325, P<0.05; evenness F2,9=13.77, P<0.05), matching the higher genotypic diversity observed for coral–Symbiodinium assemblages in this habitat (Bongaerts et al., 2010). Owing to the absence of identical host–Symbiodinium assemblages at all depth habitats, it was not possible to compare the microbial communities from ‘Back Reef’ corals with those of the ‘Upper Slope’ and ‘Deep Slope’ habitats, without it being confounded by coral–Symbiodinium genotype.
Microbial communities in the ‘Upper Slope’ were dominated by ribotypes related to Endozoicomonas (OTU 69 and 71, Figure 2), whereas microbial communities in the ‘Deep Slope’ are characterised by members of the Rhodobacteraceae (for example, OTU 25, 28 and 51; Figure 2). Of particular interest are OTU 25 and 51 which are closely related to known algal pathogens that cause bleaching and mortality of both macro- and microalgae (Imai et al., 2006; Case et al., 2011; Fernandes et al., 2011). Colonies of both the ‘Back Reef’ and ‘Deep Slope’ habitats at all locations were observed to be paler in colour than those of the ‘Upper Slope’ habitats (Supplementary Figure S2). Physiological parameters for the corals of the three depth habitats at Yonge Reef measured in a previous study found that they also had corresponding lower concentrations of areal Chlorophyll a and lower net photosynthesis (Bongaerts et al., 2011a). OTU 25 and 51 are also closely related to bacteria identified, associated with the disease lesions of White Plague Type II in the reef-building coral Montastraea faveolata (Sunagawa et al., 2009), which is characterised by an advancing bleached zone in front of the lesion boundary during a disease outbreak following a mass bleaching event (Richardson et al., 1998). The presence of potential pathogens and presentation of similar associated symptoms suggests that the health state of the ‘Deep Slope’ and ‘Back Reef’ colonies may be sub-optimal.
Although it is not possible at this stage to determine the depth-related factors responsible for the differences in microbial communities of ‘Deep Slope’ and ‘Upper Slope’ S. hystrix colonies, one explanation may be the influence of differing light levels on photosynthate production (Crossland, 1987) and the sensitivity of microbial communities to their chemical microenvironment (Ritchie and Smith, 1995a, 1995b; Ritchie, 2006; Raina et al., 2010). The lipid content of coral tissues is mainly derived from photosynthesis by the Symbiodinium (Kellogg and Patton, 1983; Harland et al., 1993) which varies with light levels, and therefore also depth (Oku et al., 2003). This photosynthetic source is assimilated into mucus by the host and contains high concentrations of proteins, polysaccharides and lipids, making it an ideal environment for microbes (Ducklow and Mitchell, 1979; Ferrier-Pages et al., 2000; Wild et al., 2004b). Differences in photosynthesis due to depth or photosynthetic potential due to the presence of photosymbiont pathogens may therefore result in changes in the condition of the coral tissues and surface mucus layer, consequently further influencing other members of the associated microbial communities (Crossland, 1987; Benlloch et al., 1995; Ikeda and Miyachi, 1995; Schäfer et al., 2001; Cooper et al., 2011; Tremblay et al., 2011; Nelson et al., 2013). This reduced function in photosynthesis in ‘Back Reef’ and ‘Deep Slope’ colonies, coupled with differing light levels would therefore result in a difference in the availability of carbon and other metabolites (Crossland, 1987; Ikeda and Miyachi, 1995; Oku et al., 2003) to the associated microbial communities. The coral microbiomes may therefore be subject to similar environmental factors such as temperature and irradiance that result in depth specificity seen in Symbiodinium-type zonation (Baker, 2003; Stat et al., 2008b).
Conclusion
It is well established that diverse microbial communities inhabit adult corals (Williams et al., 1987; Santavy, 1995; Bythell et al., 2002; Lesser et al., 2004) and have a key role in the health of their host (Koh, 1997; Castillo et al., 2001; Ritchie, 2006; Kvennefors et al., 2010; Rypien et al., 2010; Sharp et al., 2010). However, there is continuing debate as to which factors predominantly influence these communities. This is the first study to examine the differences in the composition of bacterial and archaeal communities associated with a scleractinian coral with respect to both the host coral genotype, their Symbiodinium subclade and their habitat type (location and depth). The results of this study supports those of others that habitat is the overall controlling factor of the coral microbiome and not the other main members of the holobiont. However, it must be considered that teasing apart physiological factors such as photosynthesis from environmental ones is hard as they often go hand-in-hand. Distinct changes with depth from Endozoicomonas-dominated communities to a community dominated by potential pathogens were also observed. Although it is beyond the scope of this study to determine the mechanisms behind these changes, it provides further evidence that members of the Endozoicomonas provide a beneficial function to the coral holobiont (Bourne et al., 2008; Ainsworth and Hoegh-Guldberg, 2009; Kvennefors et al., 2012; Bayer et al., 2013). Their high densities and close physical relationship to the Symbiodinium within the coral endoderm identified in previous studies and the strong negative correlation to the occurrence of bleaching pathogens and associated visible signs of coral bleaching found here suggests that they may be providing direct protection to the photosymbionts. Coupled with their intimate relationship with corals worldwide, this bacterial endosymbiont appears to be a vital member of the coral holobiont, possibly coevolving with the coral host and Symbiodinium, making them integral to the success of the coral holobiont system, and therefore warrants further investigation.
References
Ainsworth TD, Hoegh-Guldberg O . (2009). Bacterial communities closely associated with coral tissues vary under experimental and natural reef conditions and thermal stress. Aqua Biol 4: 289–296.
Anthony KRN, Fabricius KE . (2000). Shifting roles of heterotrophy and autotrophy in coral energetics under varying turbidity. J Exp Mar Biol Ecol 252: 221–253.
Apprill A, Marlow HQ, Martindale MQ, Rappe MS . (2009). The onset of microbial associations in the coral Pocillopora meandrina. ISME J 3: 685–699.
Apprill A, Hughen K, Mincer T . (2013). Major similarities in the bacterial communities associated with lesioned and healthy Fungiidae corals. Environ Microbiol 15: 2063–2072.
Baker AC . (2003). Flexibility and specificity in coral-algal symbiosis: diversity, ecology, and biogeography of Symbiodinium. Annu Rev Ecol Evol S 34: 661–689.
Balser TC, McMahon KD, Bart D, Bronson D, Coyle DR, Craig N et al. (2006). Bridging the gap between micro- and macro-scale perspectives on the role of microbial communities in global change ecology. Plant Soil 289: 59–70.
Banin E, Israely T, Kushmaro A, Loya Y, Orr E, Rosenberg E . (2000). Penetration of the coral-bleaching bacterium Vibrio shiloi into Oculina patagonica. Appl Environ Microbiol 66: 3031–3036.
Bayer T, Neave MJ, Alsheikh-Hussain A, Aranda M, Yum LK, Mincer T et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas Bacteria. Appl Environ Microbiol 79: 4759–4762.
Becker PT, Gillan DC, Eeckhaut I . (2009). Characterization of the bacterial community associated with body wall lesions of Tripneustes gratilla (Echinoidea) using culture-independent methods. J Invertebr Pathol 100: 127–130.
Beman JM, Roberts KJ, Wegley L, Rohwer F, Francis CA . (2007). Distribution and diversity of archaeal ammonia monooxygenase genes associated with corals. Appl Environ Microbiol 73: 5642–5647.
Benlloch S, RodriguezValera F, MartinezMurcia AJ . (1995). Bacterial diversity in two coastal lagoons deduced from 16S rDNA PCR amplification and partial sequencing. FEMS Microbiol Ecol 18: 267–279.
Berga M, Szekely AJ, Langenheder S . (2012). Effects of disturbance intensity and frequency on bacterial community composition and function. PLoS One 7: e36959.
Berkelmans R, Oliver JK . (1999). Large-scale bleaching of corals on the Great Barrier Reef. Coral Reefs 18: 55–60.
Berkelmans R, De'ath G, Kininmonth S, Skirving WJ . (2004). A comparison of the 1998 and 2002 coral bleaching events on the Great Barrier Reef: spatial correlation, patterns, and predictions. Coral Reefs 23: 74–83.
Bongaerts P, Riginos C, Ridgway T, Sampayo EM, van Oppen MJH, Englebert N et al. (2010). Genetic divergence across habitats in the widespread coral Seriatopora hystrix and its associated Symbiodinium. PLoS One 5: e10871.
Bongaerts P, Riginos C, Hay KB, van Oppen MJH, Hoegh-Guldberg O, Dove S . (2011a). Adaptive divergence in a scleractinian coral: physiological adaptation of Seriatopora hystrix to shallow and deep reef habitats. BMC Evol Biol 11: 303.
Bongaerts P, Sampayo EM, Bridge TCL, Ridgway T, Vermeulen F, Englebert N et al. (2011b). Symbiodinium diversity in mesophotic coral communities on the Great Barrier Reef: a first assessment. Mar Ecol Prog Ser 439: 117–126.
Bourne D, Iida Y, Uthicke S, Smith-Keune C . (2008). Changes in coral-associated microbial communities during a bleaching event. ISME J 2: 350–363.
Bourne DG, Munn CB . (2005). Diversity of bacteria associated with the coral Pocillopora damicornis from the Great Barrier Reef. Environ Microbiol 7: 1162–1174.
Bragg L, Stone G, Imelfort M, Hugenholtz P, Tyson GW . (2012). Fast, accurate error-correction of amplicon pyrosequences using Acacia. Nat Methods 9: 425–426.
Buckling A, Kassen R, Bell G, Rainey PB . (2000). Disturbance and diversity in experimental microcosms. Nature 408: 961–964.
Bythell JC, Barer MR, Cooney RP, Guest JR, O'Donnell AG, Pantos O et al. (2002). Histopathological methods for the investigation of microbial communities associated with disease lesions in reef corals. Lett Appl Microbiol 34: 359–364.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336.
Case RJ, Longford SR, Campbell AH, Low A, Tujula N, Steinberg PD et al. (2011). Temperature induced bacterial virulence and bleaching disease in a chemically defended marine macroalga. Environ Microbiol 13: 529–537.
Castillo I, Lodeiros C, Nunez M, Campos I . (2001). In vitro evaluation of antibacterial substances produced by bacteria isolated from different marine organisms. Rev Biol Trop 49: 1213–1221.
Ceh J, Raina J-B, Soo RM, van Keulen M, Bourne DG . (2012). Coral-bacterial communities before and after a coral mass spawning event on Ningaloo Reef. PLoS One 7: e36920.
Chen C-P, Tseng C-H, Chen CA, Tang S-L . (2011). The dynamics of microbial partnerships in the coral Isopora palifera. ISME J 5: 728–740.
Cooney RP, Pantos O, Le Tissier MDA, Barer MR, O'Donnell AG, Bythell JC . (2002). Characterization of the bacterial consortium associated with black band disease in coral using molecular microbiological techniques. Environ Microbiol 4: 401–413.
Cooper TF, Lai M, Ulstrup KE, Saunders SM, Flematti GR, Radford B et al. (2011). Symbiodinium genotypic and environmental controls on lipids in reef-building corals. PLoS One 6: e201434.
Crossland CJ . (1987). In situ release of mucus and DOC-lipid from the corals Acropora variabilis and Stylophora pistillata in different light regimes. Coral Reefs 6: 35–42.
De'ath G, Fabricius K . (2010). Water quality as a regional driver of coral biodiversity and macroalgae on the Great Barrier Reef. Ecol Appl 20: 840–850.
Dennis PG, Guo K, Imelfort M, Jensen P, Tyson GW, Rabaey K . (2013). Spatial uniformity of microbial diversity in a continuous bioelectrochemical system. Bioresour Technol 129: 599–605.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dinsdale EA, Pantos O, Smriga S, Edwards RA, Angly F, Wegley L et al. (2008). Microbial ecology of four coral atolls in the Northern Line Islands. PLoS One 3: e1584.
Douglas AE . (1998). Host benefit and the evolution of specialization in symbiosis. Heredity 81: 599–603.
Ducklow H, Mitchell R . (1979). Bacterial populations and adaptations in the mucus layers on living corals. Limnol Oceanogr 24: 715–725.
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R . (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27: 2194–2200.
Fabricius KE, Logan M, Weeks S, Brodie J . (2014). The effects of river run-off on water clarity across the central Great Barrier Reef. Mar Pollut Bull 84: 191–200.
Fernandes N, Case RJ, Longford SR, Seyedsayamdost MR, Steinberg PD, Kjelleberg S et al. (2011). Genomes and virulence factors of novel bacterial pathogens causing bleaching disease in the marine red alga Delisea pulchra. PLoS One 6: e27387.
Ferrier-Pages C, Leclercq N, Jaubert J, Pelegri SP . (2000). Enhancement of pico- and nanoplankton growth by coral exudates. Aqua Micro Ecol 21: 203–209.
Frias-Lopez J, Zerkle AL, Bonheyo GT, Fouke BW . (2002). Partitioning of bacterial communities between seawater and healthy, black band diseased, and dead coral surfaces. Appl Environ Microbiol 68: 2214–2228.
Furby KA, Apprill A, Cervino JM, Ossolinski JE, Hughen KA . (2014). Incidence of lesions on Fungiidae corals in the eastern Red Sea is related to water temperature and coastal pollution. Mar Env Res 98: 29–38.
Guppy R, Bythell JC . (2006). Environmental effects on bacterial diversity in the surface mucus layer of the reef coral Montastraea faveolata. Mar Ecol Prog Ser 328: 133–142.
Hansson L, Agis M, Maier C, Weinbauer MG . (2009). Community composition of bacteria associated with cold-water coral Madrepora oculata: within and between colony variability. Mar Ecol Prog Ser 397: 89–102.
Harland AD, Navarro JC, Davies PS, Fixter LM . (1993). Lipids of some Caribbean and Red Sea corals: total lipid, wax esters, triglycerides and fatty acids. Mar Biol 117: 113–117.
Hong M-J, Yu Y-T, Chen CA, Chiang P-W, Tang S-L . (2009). Influence of species specificity and other factors on bacteria associated with the coral Stylophora pistillata in Taiwan. Appl Environ Microbiol 75: 7797–7806.
Ikeda Y, Miyachi S . (1995). Carbon dioxide fixation by photosynthesis and calcification for a solitary coral, Fungia sp. Bulletin Inst Oceangra 14: 61–67.
Imai I, Fujimaru D, Nishigaki T, Kurosaki M, Sugita H . (2006). Algicidal bacteria isolated from the surface of seaweeds from the coast of Osaka Bay in the Seto Inland Sea, Japan. Afr J Mar Sci 28: 319–323.
Jessen C, Lizcano JFV, Bayer T, Roder C, Aranda M, Wild C et al. (2013). In-situ effects of eutrophication and overfishing on physiology and bacterial diversity of the Red Sea coral Acropora hemprichii. PLoS One 8: e62091.
Jiang L, Patel SN . (2008). Community assembly in the presence of disturbance: a microcosm experiment. Ecology 89: 1931–1940.
Johnston IS, Rohwer F . (2007). Microbial landscapes on the outer tissue surfaces of the reef-building coral Porites compressa. Coral Reefs 26: 375–383.
Kellogg RB, Patton JS . (1983). Lipid droplets, medium of energy exchange in the symbiotic anemone Condylactis gigantea: a model coral polyp. Mar Biol 75: 137–149.
Kimes NE, Van Nostrand JD, Weil E, Zhou J, Morris PJ . (2010). Microbial functional structure of Montastraea faveolata, an important Caribbean reef-building coral, differs between healthy and yellow-band diseased colonies. Environ Microbiol 12: 541–556.
Klaus JS, Janse I, Heikoop JM, Sanford RA, Fouke BW . (2007). Coral microbial communities, zooxanthellae and mucus along gradients of seawater depth and coastal pollution. Environ Microbiol 9: 1291–1305.
Koh EGL . (1997). Do scleractinian corals engage in chemical warfare against microbes? J Chem Ecol 23: 379–398.
Koren O, Rosenberg E . (2008). Bacteria associated with the bleached and cave coral Oculina patagonica. Microb Ecol 55: 523–529.
Kurahashi M, Yokota A . (2007). Endozoicomonas elysicola gen. nov., sp nov., a gamma-proteobacterium isolated from the sea slug Elysia ornata. Syst Appl Microbiol 30: 202–206.
Kvennefors ECE, Sampayo EM, Ridgway T, Barnes AC, Hoegh-Guldberg O . (2010). Bacterial communities of two ubiquitous Great Barrier Reef corals reveals both site- and species-specificity of common bacterial associates. PLoS One 5: e10401.
Kvennefors ECE, Sampayo E, Kerr C, Vieira G, Roff G, Barnes AC . (2012). Regulation of bacterial communities through antimicrobial activity by the coral holobiont. Microb Ecol 63: 605–618.
LaJeunesse TC, Loh WKW, van Woesik R, Hoegh-Guldberg O, Schmidt GW, Fitt WK . (2003). Low symbiont diversity in southern Great Barrier Reef corals, relative to those of the Caribbean. Limnol Oceanogr 48: 2046–2054.
LaJeunesse TC, Thornhill DJ, Cox EF, Stanton FG, Fitt WK, Schmidt GW . (2004). High diversity and host specificity observed among symbiotic dinoflagellates in reef coral communities from Hawaii. Coral Reefs 23: 596–603.
LaJeunesse TC . (2005). ‘Species’ radiations of symbiotic dinoflagellates in the Atlantic and Indo-Pacific since the Miocene-Pliocene transition. Mol Biol Evol 22: 570–581.
LaJeunesse TC, Pettay DT, Sampayo EM, Phongsuwan N, Brown B, Obura DO et al. (2010). Long-standing environmental conditions, geographic isolation and host-symbiont specificity influence the relative ecological dominance and genetic diversification of coral endosymbionts in the genus Symbiodinium. J Biogeogr 37: 785–800.
Leggat W, Hoegh-Guldberg O, Dove S, Yellowlees D . (2007). Analysis of an EST library from the dinoflagellate (Symbiodinium sp.) symbiont of reef-building corals. J Phycol 43: 1010–1021.
Lema KA, Willis BL, Bourne DG . (2012). Corals form characteristic associations with symbiotic nitrogen-fixing bacteria. Appl Environ Microbiol 78: 3136–3144.
Lesser MP, Mazel CH, Gorbunov MY, Falkowski PG . (2004). Discovery of symbiotic nitrogen-fixing cyanobacteria in corals. Science 305: 997–1000.
Lesser MP, Falcon LI, Rodriguez-Roman A, Enriquez S, Hoegh-Guldberg O, Iglesias-Prieto R . (2007). Nitrogen fixation by symbiotic cyanobacteria provides a source of nitrogen for the scleractinian coral Montastraea cavernosa. Mar Ecol Prog Ser 346: 143–152.
Lins-de-Barros MM, Vieira RP, Cardoso AM, Monteiro VA, Turque AS, Silveira CB et al. (2010). Archaea, bacteria, and algal plastids associated with the reef-building corals Siderastrea stellata and Mussismilia hispida from Búzios, South Atlantic Ocean, Brazil. Microb Ecol 59: 523–532.
Littman R, Willis BL, Bourne DG . (2011). Metagenomic analysis of the coral holobiont during a natural bleaching event on the Great Barrier Reef. Environ Microbiol Rep 3: 651–660.
Littman RA, Willis BL, Bourne DG . (2009a). Bacterial communities of juvenile corals infected with different Symbiodinium (dinoflagellate) clades. Mar Ecol Prog Ser 389: 45–59.
Littman RA, Willis BL, Pfeffer C, Bourne DG . (2009b). Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the Great Barrier Reef. FEMS Microbiol Ecol 68: 152–163.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhu kumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Meikle P, Richards GN, Yellowlees D . (1988). Structural investigations on the mucus from six species of coral. Mar Biol 99: 187–193.
Mouchka ME, Hewson I, Harvell CD . (2010). Coral-associated bacterial assemblages: current knowledge and the potential for climate-driven impacts. Integr Comp Biol 50: 662–674.
Muscatine L, Porter JW . (1977). Coral Reefs: mutualistic symbioses adapted to nutrient-poor environments. Bioscience 27: 454–460.
Nelson CE, Goldberg SJ, Kelly LW, Haas AF, Smith JE, Rohwer F et al. (2013). Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J 7: 962–979.
Oku H, Yamashiro H, Onaga K, Sakai K, Iwasaki H . (2003). Seasonal changes in the content and composition of lipids in the coral Goniastrea aspera. Coral Reefs 22: 83–85.
Olson ND, Ainsworth TD, Gates RD, Takabayashi M . (2009). Diazotrophic bacteria associated with Hawaiian Montipora corals: diversity and abundance in correlation with symbiotic dinoflagellates. J Exp Mar Biol Ecol 371: 140–146.
Paul JH, DeFlaun MF, Jeffery WH . (1986). Elevated levels of microbial activity in the coral surface monolayer. Mar Ecol Prog Ser 33: 29–40.
Pernice M, Meibom A, Van Den Heuvel A, Kopp C, Domart-Coulon I, Hoegh-Guldberg O et al. (2012). A single-cell view of ammonium assimilation in coral-dinoflagellate symbiosis. ISME J 6: 1314–1324.
Peters EC . (1984). A survey of cellular reactions to environmental-stress and disease in Caribbean scleractinian corals. Helgolander Meeresuntersuchungen 37: 113–137.
Raina J-B, Dinsdale EA, Willis BL, Bourne DG . (2010). Do the organic sulfur compounds DMSP and DMS drive coral microbial associations? Trends Microbiol 18: 101–108.
Richardson LL, Goldberg WM, Carlton RG, Halas JC . (1998). Coral disease outbreak in the Florida keys: plague type II. Rev Biol Trop 46: 187–198.
Ritchie KB, Smith GW . (1995a). Preferential carbon utilization by surface bacterial communities from water mass, normal, and white-band diseased Acropora cervicornis. Mol Mar Biol Biotech 4: 345–352.
Ritchie KB, Smith GW . (1995b). Carbon-source utilization patterns of coral-associated marine heterotrophs. J Mar Biotechnol 3: 105–107.
Ritchie KB, Smith GW . (2004). Microbial communities of coral surface mucopolysaccharide layers. In: Rosenberg E, Loya Y (eds) Coral Health and Disease. Springer-Verlag: Berlin, Germany, pp 259–263.
Ritchie KB . (2006). Regulation of microbial populations by coral surface mucus and mucus-associated bacteria. Mar Ecol Prog Ser 322: 1–14.
Rohwer F, Breitbart M, Jara J, Azam F, Knowlton N . (2001). Diversity of bacteria associated with the Caribbean coral Montastraea franksi. Coral Reefs 20: 85–91.
Rohwer F, Seguritan V, Azam F, Knowlton N . (2002). Diversity and distribution of coral-associated bacteria. Mar Ecol Prog Ser 243: 1–10.
Rypien KL, Ward JR, Azam F . (2010). Antagonistic interactions among coral-associated bacteria. Environ Microbiol 12: 28–39.
Santavy DL . (1995). The diversity of microorganisms associated with marine invertebrates and their roles in the maintenance of ecosystems. In: Allsopp D, Colwell RR, Hawksworth DL eds Microbial Diversity and Ecosystem Function. CAB International: Wallingford, WI, USA, pp 211–229.
Santavy DL, Peters EC . (1997). Microbial pests: Coral disease in the western Atlantic. Proc 8th Int Coral Reef Symp 1: 607–612.
Santiago-Vazquez LZ, Brueck TB, Brueck WM, Duque-Alarcon AP, McCarthy PJ, Kerr RG . (2007). The diversity of the bacterial communities associated with the azooxanthellate hexacoral Cirrhipathes lutkeni. ISME J 1: 654–659.
Schäfer H, Bernard L, Courties C, Lebaron P, Servais P, Pukall R et al. (2001). Microbial community dynamics in Mediterranean nutrient-enriched seawater mesocosms: changes in the genetic diversity of bacterial populations. FEMS Microbiol Ecol 34: 243–253.
Schöttner S, Wild C, Hoffmann F, Boetius A, Ramette A . (2012). Spatial scales of bacterial diversity in cold-water coral reef ecosystems. PLoS One 7: e32093.
Schuett C, Doepke H, Grathoff A, Gedde M . (2007). Bacterial aggregates in the tentacles of the sea anemone Metridium senile. Helgoland Mar Res 61: 211–216.
Sekar R, Kaczmarsky LT, Richardson LL . (2009). Effect of freezing on PCR amplification of 16S rRNA genes from microbes associated with black band disease of corals. Appl Environ Microbiol 75: 2581–2584.
Sharp KH, Ritchie KB, Schupp PJ, Ritson-Williams R, Paul VJ . (2010). Bacterial acquisition in juveniles of several broadcast spawning coral species. PLoS One 5: e10898.
Sharp KH, Distel D, Paul VJ . (2012). Diversity and dynamics of bacterial communities in early life stages of the Caribbean coral Porites astreoides. ISME J 6: 790–801.
Shashar N, Cohen Y, Loya Y, Sar N . (1994). Nitrogen-fixation (acetylene-reduction) in stony corals: evidence for coral-bacteria interactions. Mar Ecol Prog Ser 111: 259–264.
Siboni N, Ben-Dov E, Sivan A, Kushmaro A . (2008). Global distribution and diversity of coral-associated Archaea and their possible role in the coral holobiont nitrogen cycle. Environ Microbiol 10: 2979–2990.
Siboni N, Ben-Dov E, Sivan A, Kushmaro A . (2012). Geographic specific coral-associated ammonia-oxidizing Archaea in the Northern Gulf of Eilat (Red Sea). Microb Ecol 64: 18–24.
Silverstein RN, Correa AMS, LaJeunesse TC, Baker AC . (2011). Novel algal symbiont (Symbiodinium spp.) diversity in reef corals of Western Australia. Mar Ecol Prog Ser 422: 63–75.
Simpson EH . (1949). Measurement of diversity. Nature 163: 688–688.
Sorokin YI . (1973). Trophic role of bacteria in ecosystem of coral reef. Nature 242: 415–417.
Speck MD, Donachie SP . (2012). Widespread Oceanospirillaceae bacteria in Porites spp. J Mar Biol 2012: 7.
Stat M, Loh WKW, Hoegh-Guldberg O, Carter DA . (2008a). Symbiont acquisition strategy drives host-symbiont associations in the southern Great Barrier Reef. Coral Reefs 27: 763–772.
Stat M, Morris E, Gates RD . (2008b). Functional diversity in coral-dinoflagellate symbiosis. Proc Natl Acad Sci USA 105: 9256–9261.
Sunagawa S, DeSantis TZ, Piceno YM, Brodie EL, DeSalvo MK, Voolstra CR et al. (2009). Bacterial diversity and white plague disease-associated community changes in the Caribbean coral Montastraea faveolata. ISME J 3: 512–521.
Sunagawa S, Woodley CM, Medina M . (2010). Threatened corals provide underexplored microbial habitats. PLoS One 5: e9554.
Tremblay P, Weinbauer MG, Rottier C, Guerardel Y, Nozais C, Ferrier-Pages C . (2011). Mucus composition and bacterial communities associated with the tissue and skeleton of three scleractinian corals maintained under culture conditions. J Mar Biol Assoc UK 91: 649–657.
Veron JEN, Pichon M . (1976) Marine Science Monograph Series 1: Scleractinia of Eastern Australia, Part 1. Families Thamnasteriidae, Astrocoeniidae, and Pocilloporidae. Australian Institute of Marine Science: Townsville, QLD, Australia.
Veron JEN . (2000) Corals of the World vol. 1–3. Australian Institute of Marine Science: Townsville, QLD, Australia.
Wegley L, Yu YN, Breitbart M, Casas V, Kline DI, Rohwer F . (2004). Coral-associated archaea. Mar Ecol Prog Ser 273: 89–96.
Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F . (2007). Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 9: 2707–2719.
Wild C, Huettel M, Klueter A, Kremb SG, Rasheed MYM, Jorgensen BB . (2004a). Coral mucus functions as an energy carrier and particle trap in the reef ecosystem. Nature 428: 66–70.
Wild C, Rasheed M, Werner U, Franke U, Johnstone R, Huettel M . (2004b). Degradation and mineralization of coral mucus in reef environments. Mar Ecol Prog Ser 267: 159–171.
Wild C, Naumann M, Niggl W, Haas A . (2010a). Carbohydrate composition of mucus released by scleractinian warm- and cold-water reef corals. Aqua Biol 10: 41–45.
Wild C, Niggl W, Naumann MS, Haas AF . (2010b). Organic matter release by Red Sea coral reef organisms-potential effects on microbial activity and in situ O2 availability. Mar Ecol Prog Ser 411: 61–71.
Williams WM, Viner AB, Broughton WJ . (1987). Nitrogen-fixation (acetylene-reduction) associated with the living coral Acropora variabilis. Mar Biol 94: 531–535.
Yakimov MM, Cappello S, Crisafi E, Tursi A, Savini A, Corselli C et al. (2006). Phylogenetic survey of metabolically active microbial communities associated with the deep-sea coral Lophelia pertusa from the Apulian plateau, Central Mediterranean Sea. Deep-Sea Res Pt I 53: 62–75.
Yang CS, Chen MH, Arun AB, Chen CA, Wang JT, Chen WM . (2010). Endozoicomonas montiporae sp nov., isolated from the encrusting pore coral Montipora aequituberculata. Int J Syst Evol Microbiol 60: 1158–1162.
Acknowledgements
This study was conducted as part of the ‘Catlin Seaview Survey’ funded by Catlin Group Limited, with support from the Australian Research Council (ARC) Centre of Excellence for Coral Reef Studies and the Global Change Institute. GWT is supported by an ARC Queen Elizabeth II fellowship (DP1093175).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article

Cite this article
Pantos, O., Bongaerts, P., Dennis, P. et al. Habitat-specific environmental conditions primarily control the microbiomes of the coral Seriatopora hystrix. ISME J 9, 1916–1927 (2015). https://doi.org/10.1038/ismej.2015.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.3
This article is cited by
-
Microbiome changes in Sinularia spp. soft corals relative to health status
Marine Biology (2024)
-
Metagenomics reveals the habitat specificity of biosynthetic potential of secondary metabolites in global food fermentations
Microbiome (2023)
-
Diversity of the Pacific Ocean coral reef microbiome
Nature Communications (2023)
-
Differential responses of bacterial communities in coral tissue and mucus to bleaching
Coral Reefs (2022)
-
Naturally occurring fire coral clones demonstrate a genetic and environmental basis of microbiome composition
Nature Communications (2021)
