
Abstract
Plant–microbe interactions in the rhizosphere have important roles in biogeochemical cycling, and maintenance of plant health and productivity, yet remain poorly understood. Using RNA-based metatranscriptomics, the global active microbiomes were analysed in soil and rhizospheres of wheat, oat, pea and an oat mutant (sad1) deficient in production of anti-fungal avenacins. Rhizosphere microbiomes differed from bulk soil and between plant species. Pea (a legume) had a much stronger effect on the rhizosphere than wheat and oat (cereals), resulting in a dramatically different rhizosphere community. The relative abundance of eukaryotes in the oat and pea rhizospheres was more than fivefold higher than in the wheat rhizosphere or bulk soil. Nematodes and bacterivorous protozoa were enriched in all rhizospheres, whereas the pea rhizosphere was highly enriched for fungi. Metabolic capabilities for rhizosphere colonisation were selected, including cellulose degradation (cereals), H2 oxidation (pea) and methylotrophy (all plants). Avenacins had little effect on the prokaryotic community of oat, but the eukaryotic community was strongly altered in the sad1 mutant, suggesting that avenacins have a broader role than protecting from fungal pathogens. Profiling microbial communities with metatranscriptomics allows comparison of relative abundance, from multiple samples, across all domains of life, without polymerase chain reaction bias. This revealed profound differences in the rhizosphere microbiome, particularly at the kingdom level between plants.
Similar content being viewed by others
Introduction
Interactions between plants and microbes in the rhizosphere are of global importance to biogeochemical cycling (Philippot et al., 2009), plant health and productivity (Bloemberg and Lugtenberg, 2001). Colonisation of the rhizosphere, the region of soil influenced by plant roots, is necessary for both plant pathogens and plant growth-promoting rhizobacteria. The latter aid plants by providing nutrients, modulating growth, defending against diseases (Lugtenberg and Kamilova, 2009) and contributing to disease-suppressive soils (Mendes et al., 2011). Many plant-associated microbes are known and well-studied, including the symbiotic nitrogen-fixing Rhizobium leguminosarum (Young et al., 2006), and both beneficial and pathogenic Pseudomonas spp. (Feil et al., 2005; Paulsen et al., 2005). Also, association of mycorrhizal fungi with most land plants is fundamental to acquisition of mineral nutrients such as phosphate (Bonfante, 2010). However, little is known about how these organisms interact at the community level.
Every gram of soil is estimated to contain in excess of 50 000 species of bacteria (Roesch et al., 2007), the vast majority of which are uncultured (Handelsman, 2004). Sequencing of polymerase chain reaction (PCR)-amplified 16S rDNA has been extensively used to examine rhizosphere bacterial communities of various plants, and recently high-throughput pyrosequencing (Margulies et al., 2005) has revealed these communities in previously unobtainable detail. Plants studied include the important crop potato (Solanum tuberosum) (Inceoglu et al., 2011), plants of the Antarctic (Teixeira et al., 2010) and recently the model dicot Arabidopsis thaliana (Bulgarelli et al., 2012; Lundberg et al., 2012). However, PCR amplification of genomic DNA is inherently biased by primer design (Hong et al., 2009; Pinto and Raskin, 2012) and is limited to the targeted division of life (Bacteria, Archaea, Eukarya or a smaller taxonomic group). Studies to date have largely focused on either bacteria or fungi, often neglecting other eukaryotes and archaea.
High-throughput sequencing has also enabled the use of metagenomic strategies where total genomic DNA from the rhizosphere is sequenced (Tett et al., 2012). While encompassing all domains of life and indicating the metabolic potential of a microbiome, a metagenome contains relatively few rRNA genes, reducing the strength of taxonomic assignments. Metatranscriptomics, where total RNA from the environment is sequenced, reveals active community members and metabolic pathways (Urich et al., 2008). Many applications of metatranscriptomics are focussed on the latter but have a significant challenge in the requirement for enrichment of mRNA (Stewart et al., 2010; Yi et al., 2011). However, the dominance of rRNA in a metatranscriptomic sample allows robust assessment of the entire microbiome, without prior selection of taxonomic groups for study. This is technically much less challenging than enrichment of mRNA, avoids PCR bias and can be carried out straightforwardly on multiple samples.
In this study, comparative metatranscriptomics was used to study the rhizosphere microbiomes of three crop plants grown in the same soil: wheat (Triticum aestivum), a major world food staple; oat (Avena strigosa), a cereal that produces anti-fungal avenacins (Maizel et al., 1964); and pea (Pisum sativum), a widely grown crop legume nodulated by N2-fixing bacteria R. leguminosarum. In addition, we compared the rhizosphere microbiome of the wild-type oat with that of an avenacin-deficient mutant, sad1 (Haralampidis et al., 2001). Avenacins are triterpenoid saponins that protect oat from root pathogens (Papadopoulou et al., 1999) including Gaeumannomyces graminis, the causative agent of take-all (Osbourn et al., 1994). Although metatranscriptomic analysis of total rRNA has been used to profile microbial communities in soil (Urich et al., 2008) and oceans (Ottesen et al., 2011; Shi et al., 2011), this is, to our knowledge, the first application of it to study the rhizosphere microbiomes of important crop plants.
Materials and methods
Plant growth and sampling
Soil was collected from an agricultural field near Norwich, UK (52°62′29′′N, 1°21′81′′E) in March 2009. A chemical analysis was performed with soil from the same site by Macaulay Soils (Aberdeen, UK). Soil pH was 7.49, with an organic matter content of 2.3%. Nitrate (NO3−), phosphate (PO43−), potassium (K+) and magnesium (Mg2+) concentrations were 149, 171, 184, and 60 mg kg−1, respectively. These are considered high for nitrate and phosphate, moderate for potassium and low for magnesium.
Plants used were wheat (T. aestivum var. Paragon), diploid oat (A. strigosa accession S75) and pea (P. sativum var. Avola). An oat mutant deficient in avenacin production (sad1) was also used (Haralampidis et al., 2001). Seeds were surface sterilised with 5% hypochlorite solution (wheat and oat) or 95% ethanol and 2% hypochlorite solution (pea), and germinated on moist filter paper for 2–3 days at room temperature in the dark. Seedlings were planted in 500 ml closed pots and grown for 4 weeks in a glasshouse, with distilled water added as necessary. Plant-free pots were also set up as controls, and given the same treatment as plants. The sad1 oat mutant showed the same growth behaviour as the wild type, and despite the high nitrogen levels in the soil, the pea plants were nodulated. Plants were harvested and loosely attached soil was discarded by shaking. Stems were removed, and dry roots were vortexed in a 50 ml tube to remove soil closely adhering to the root, which we consider to be rhizosphere soil. Approximately 1 g soil was added to a bead tube from the RNA PowerSoil Total RNA Isolation kit (MoBio, Carlsbad, CA, USA), and RNA extracted according to the manufacturer’s instructions. Yield and quality were determined using an Experion bioanalyser (Bio-Rad, Hercules, CA, USA).
RNA processing and sequencing
Total RNA samples were treated with TurboDNase (Ambion, Austin, TX, USA), cleaned using the RNeasy Minikit (Qiagen, Valencia, CA, USA) and converted to cDNA using strand displacement amplification with the Complete Whole Transcriptome Amplification kit WTA2 (Rubicon Genomics, Ann Arbor, MI, USA). This method is reliable, straightforward and is less expensive than alternative cDNA generation kits. It also requires very little (25 ng) input RNA, which is an important advantage when dealing with environmental samples. The synthesis reaction was purified using the QIAquick PCR Purification kit (Qiagen), and the resulting cDNA assessed with a high-sensitivity Agilent 2100 bioanalyser (Agilent, Santa Clara, CA, USA). Size profiles of cDNA fragments were consistent across samples. Multiplexing and sequencing were carried out at The Genome Analysis Centre (TGAC, Norwich, UK) on two runs of a 454 GS Flx sequencer (Roche 454 Life Sciences, Branford, CT, USA). A total of 1 674 231 reads were generated for the 19 biologically independent samples (four each from soil and oat, three each from wheat and pea, five from the sad1 mutant oat line). PHRED quality scores were 32.3 and 34.3 for the two runs, with standard deviations of 8.3 and 7.7, respectively.
Bioinformatic analysis
Sequence reads were de-multiplexed and used in downstream analyses without filtering artificial replicates (Gomez-Alvarez et al., 2009). Owing to the dominance and similarity of rRNA sequences, omitting this filtering step prevents genuine biological replicates being removed, which would downweight abundant taxa. Filtering has been omitted from another metatranscriptomic study (Ottesen et al., 2011). The conserved 22 bp tail generated by WTA2 procedure for cDNA synthesis was removed using a Perl script.
Read files were used as queries against a cleaned and de-replicated (95% identity) set of sequences from the small subunit (SSU) SILVA (Pruesse et al., 2007) and RDP (Cole et al., 2009) rRNA databases using USEARCH in UBLAST mode (Edgar, 2010) followed by post-processing to extract taxonomic statistics. An E-value cutoff of 10−7 was applied, the top 100 hits were recorded in an output file, and in the process short reads (<10 bp) were discarded. Output files were uploaded into MEGAN (Huson et al., 2007) using default parameters, except Min. Support was set to 1, and top percent to 5. These parameters reduced underprediction for highly similar rRNA sequences.
A comparison file was generated for all 19 samples using absolute counts, and numbers of assigned reads per taxa were extracted for different taxonomic levels. Rhizosphere samples were inevitably contaminated with host plant material, which is indistinguishable from other plants based on 18S rRNA sequence. Reads were therefore normalised by expressing as a percentage of the total number of reads assigned in MEGAN minus any reads that were assigned to Viridiplantae (Table 1). Means were calculated for each group of samples from the same environment. An unpaired t-test was used to determine significant differences for pairwise comparisons between each of the wild-type plant rhizospheres with soil, and for the wild-type oat versus the sad1 oat mutant. Statistically significant differences were further filtered using a relative abundance cutoff of 0.01% of assigned reads for the environment in which they were more abundant. For example, a taxon statistically more abundant in the wheat rhizosphere compared with bulk soil would be ignored unless it contributed at least 0.01% of the reads assigned to the wheat rhizosphere community.
Between-classes principal component analysis (PCA) was carried out using the R package ade4 (Dray and Dufour, 2007). Before analysis, samples were divided (>100 000 reads) or multiplied (<100 000 samples) to normalise the abundance to the equivalent of 100 000 reads in MEGAN and low abundance taxa removed if the average relative abundance across all the samples was <0.01% (Figure 1) or <0.1% (Figure 2). Rarefaction analyses were performed separately on prokaryotes and eukaryotes at the phylum and genus levels for each sample using MEGAN. Data were extracted and absolute read numbers were calculated. Means for both the number of reads sampled and the number of taxa were generated for each group of samples, and then used to plot rarefaction curves.

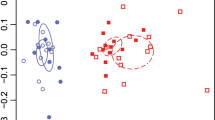
Taxonomic differences between rhizospheres as revealed by between-classes PCA. The ade4 R package (Dray and Dufour, 2007) was used to plot two principal components based on data from prokaryotic and eukaryotic communities at the phylum and genus levels. The centre of gravity for each class of rhizosphere is given by the intersection of sample lines and the ellipse covers 67% of the samples belonging to that class. The closer two ellipses appear, the greater the similarity in their community makeup. The statistical significance of the between-classes PCA, as determined by a Monte-Carlo test (n=999), gave P<0.01 in all four cases, indicating the existence of highly significant differences between the groups.

Genus level differences between major eukaryotic groups (Figure 1c) in the rhizospheres as revealed by between-classes PCA. The ade4 R package (Dray and Dufour, 2007) was used to plot two principal components based on data from eukaryotic communities at genus level. The centre of gravity for each class of rhizosphere is given by the intersection of sample lines and the ellipse covers 67% of the samples belonging to that class. The closer two ellipses appear, the greater the similarity in their community makeup. The statistical significance of the between-classes PCA, as determined by a Monte-Carlo test (n=999), gave P<0.004 for Fungi, P<0.002 for Nematoda, P<0.001 for Amoebozoa and P<0.035 for Alveolata, indicating the existence of highly significant differences between the groups.
Results and discussion
Sequencing and analysis summary
There was large variation in read output across samples (Table 1), most likely due to amplification bias of the 454 barcodes (Alon et al., 2011) introduced during the emPCR step of library preparation and small variations in input DNA levels. This is important to account for when designing multiplexed pyrosequencing experiments. By normalising data and making only relative comparisons, statistical challenges arising from this variation were avoided.
The USEARCH analysis allowed data to be processed in a few hours on a desktop computer. On average, roughly half the reads matched a sequence in the custom SSU rRNA database, and the majority of those were assigned to a taxon in MEGAN (Table 1). This is higher than previously reported in soil, where 38.3% of reads matched SSU rRNA (Urich et al., 2008). The remainder was mostly large subunit rRNA or rarely mRNA sequences. Although these can be taxonomically assigned, only sequences matching SSU rRNA were analysed further. This allowed easier comparison with published studies where PCR amplification of 16S and 18S rRNA genes has been used.
The analysed rRNA sequences were derived from full-length rRNA transcripts. Using a conservative Lowest Common Ancestor algorithm (MEGAN), false assignment of conserved sequences to low taxonomic ranks was avoided. A large proportion of reads were therefore assigned to high taxonomic ranks. Averaged across all samples, 5.5% of reads could not be classified more specifically than as cellular organisms, 81.4% were assigned to prokaryotes and 13.1% were assigned to eukaryotes. Within prokaryotes, 54.6% of reads were assigned to the phylum level, whereas only 13.5% were assigned to the genus level. For eukaryotes, 58.6% of reads were assigned to the phylum level and 37.2% were assigned to the genus level (Supplementary Figure 1). Although some reads were confidently assigned to species and strain level, this approach is less discriminating at low-level taxonomic ranks than PCR targeted to a variable region of an rRNA gene, requiring greater sequencing effort to obtain the same level of details.
Total community structure and diversity
The proportion of sequences derived from Bacteria varied between environments, ranging from 91.0% for bulk soil, 88.5% for wheat rhizosphere, 77.3% for oat rhizosphere and 73.7% for pea rhizosphere (Figure 3a). These were significantly different from bulk soil for all three rhizospheres (P=0.0465, 0.0201 and 0.0107, respectively). Eukaryotes made up 2.8% of the bulk soil community and 3.3%, 16.6% and 20.7% of the wheat, oat and pea rhizosphere communities, respectively (Figure 3a). These were only significantly different from bulk soil for oat and pea rhizospheres (P=0.0380 and 0.0167, respectively). This striking difference in relative abundance of eukaryotes demonstrates the strength of a metatranscriptomic approach, rather than PCR-based strategy, to detect the kingdom level differences between microbiomes. Archaea were consistently represented at around 0.5% (Figure 3a) for all environments, comparable to a study of soil (Urich et al., 2008).

Relative abundance of taxonomic groups in soil, wheat, wt oat, sad1 oat and pea rhizospheres. (a) All taxa at the domain level. The eukaryotes wedge excludes sequences from Viridiplantae. (b) Prokaryotic phyla, and is a subset of the Bacteria and Archaea wedges from (a). (c) Four major eukaryotic groups and is a subset of the eukaryotes wedge from (a). Both (b) and (c) only include confidently assigned reads, that is, not reads that cannot be assigned more specifically than to Bacteria, Archaea (b) or eukaryotes (c). Values are means of biological replicates, where n=3 for wheat and pea, n=4 for soil and wt oat and n=5 for sad1 oat.
Differences between microbiomes were visualised using between-classes PCA. Prokaryotic and eukaryotic communities were analysed separately, at both the phylum and genus level. At the phylum level, prokaryotic communities of oat and pea were distinct from bulk soil, whereas that of wheat was not (Figure 1). At the genus level, prokaryotic communities in all wild-type plant rhizospheres were different from soil, and more different from each other. This suggests that selection of rhizosphere microbiomes was largely plant specific. The pea rhizosphere microbiome was strikingly distinct from that of the other plants, suggesting a strong difference in the effect of a legume versus a cereal. Eukaryotic communities of bulk soil, wheat and pea were similar at the phylum level, whereas that of oat was distinct (Figure 1). At the genus level, the eukaryotic communities of wheat and bulk soil were similar, whereas oat and especially pea were different, most noticeably for Fungi and Amoebozoa (Figure 2). There was generally low variability within sample groups, although this increased at lower taxonomic levels, particularly for oat.
Rarefaction analyses of prokaryotic communities (Supplementary Figure 2) showed that the phylum level diversity of bulk soil and rhizospheres was similar, whereas at the genus level the oat rhizosphere had slightly reduced diversity compared with the other environments. Diversity of eukaryotic phyla and genera in rhizosphere samples was greater than in bulk soil. Eukaryotic diversity was surprisingly high, equalling or exceeding that of prokaryotes at the phylum level. Previous estimates of bacterial diversity suggest almost 700 different genera per gram of soil (Roesch et al., 2007), around twofold greater than that seen for our samples. However, differences in sample and analysis methods prevent a direct comparison with this study.
Highly abundant microbes in soil and rhizospheres
In bulk soil and all plant rhizospheres, the most abundant prokaryotes were the Proteobacteria, followed by Actinobacteria, Firmicutes, Acidobacteria, Planctomycetes and Bacteroidetes (Figure 3b). Some of the bulk soil and sad1 oat samples also showed high proportions of Cyanobacteria. These major groups were identified in a metatranscriptomic study of soil (Urich et al., 2008), and have been well represented in PCR-based analyses of soil (Roesch et al., 2007) and the rhizosphere (Inceoglu et al., 2011; Bulgarelli et al., 2012; Lundberg et al., 2012). In our study, the Acidimicrobiales, Actinomycetales and Bacillales contributed most to their respective phyla (Figure 4b), whereas representation of Proteobacteria was more diverse. Major contributions came from the Myxococcales and unclassified Deltaproteobacteria, as well as Alpha, Beta and Gamma subdivisions, namely the Rhizobiales, Burkholderiales and Pseudomonadales. respectively (Figure 4a). These taxa are well known for their interactions with plants and have previously been detected in soil (Janssen, 2006), rhizosphere (Lu et al., 2006) and phyllosphere (Yashiro et al., 2011) environments. At these taxonomic levels there was a high level of consistency across all environments.

Contribution of prokaryotic orders and classes to their respective phyla. Proteobacteria (a) are derived from the Proteobacteria wedge of Figure 3b, whereas other prokaryotes (b) are derived from the remaining wedges of Figure 3b. Values are means of biological replicates, where n=3 for wheat and pea, n=4 for soil and wt oat and n=5 for sad1 oat.
The majority of eukaryotic sequences were derived from Fungi and Nematoda, with some contribution from Amoebozoa and Alveolata (Figure 3c). In addition to eukaryotes being over fivefold more abundant in oat and pea rhizospheres compared with both wheat rhizosphere and soil (Figure 3a), the proportion of major eukaryotic groups was different. Nematoda were more abundant in all rhizospheres compared with soil, whereas the pea rhizosphere was highly enriched for fungi (Figure 3c).
Plant selection of microbes
Between-classes PCA of total community structure showed that plants had specific effects on their rhizospheres. The dissimilarity in community structure between a rhizosphere and bulk soil can largely be attributed to those taxa most strongly selected or depleted by the plant. Here, taxa were considered strongly selected if they were ⩾5-fold more abundant in a rhizosphere compared with the bulk soil (Supplementary Tables 1 and 2). Prokaryotic taxa most strongly selected by wheat included Dyadobacter, Fibrobacteriaceae and Verrucomicrobium. Firmicutes including Bacillus and Lysinibacillus were also enriched. Strongly enriched eukaryotes included the bacterivorous nematode Acrobeloides, and Eurotiomycete fungi. The oat rhizosphere was strongly enriched for unclassified Actinobacteridae, and less so for Comamonadaceae, Pseudomonadaceae and the photoheterotrophic Chloroflexaceae. Strongly selected eukaryotes included Euglenozoa and the Amoebozoa Glaeseria and Leptomyxa.
The pea rhizosphere was strongly enriched for Masillia, Dyadobacter, Flavobacterium and Streptomyces. Other taxa less strongly selected included known plant growth-promoting rhizobacteria Stenotrophomonas (Ryan et al., 2009) and Variovorax (Han et al., 2011). Strains of Flavobacterium and Variovorax have been isolated from soil adjacent to N2-fixing nodules of soybean (Glycine max) and were shown to oxidise H2, and promote plant growth (Maimaiti et al., 2007). Importantly, H2 is a by-product of N2 reduction by nitrogenase (Hunt and Layzell, 1993) and is thought to drive selection of plant beneficial microbes in legume rhizospheres (Dong et al., 2003). Eukaryotes strongly selected by pea included the ciliate Bresslaua, flagellate Dimastigella, and root-knot nematode Meloidogyne, in addition to numerous fungi. These included Tetracladium, Fusarium and Exophiala, which have been associated with diseased peas (Yu et al., 2012), as well as the mycorrhizal Glomeromycota. The latter is of particular significance given the shared developmental pathways of Rhizobium–legume and mycorrhizal symbioses (Stracke et al., 2002), hinting that legumes may have enhanced associations with mycorrhiza compared with non-legumes. There were increases in relative abundance of Glomeromycota in wheat and oat rhizospheres, but these differences were not statistically significant.
Selection of taxa with metabolic capabilities potentially important in rhizosphere colonisation was observed. Cellulolytic bacteria such as Fibrobacteres (Ransom-Jones et al., 2012) and Cellvibrio were selected by both wheat and oat. This suggested the presence of plant cell wall material in the rhizosphere, which is often overlooked in the context of shaping the rhizosphere microbiome (Dennis et al., 2010). The cereals also selected for unclassified Methylophilaceae, which can reduce nitrate (high in this soil) in the presence of methanol (Kalyuhznaya et al., 2009). Methanol is produced from pectin in plant cell walls (Galbally and Kirstine, 2002), and metabolism of C1 compounds is active during bacterial colonisation of the phyllosphere (Delmotte et al., 2009) and rhizosphere (Ramachandran et al., 2011; Knief et al., 2012). Enrichment of other methylotrophs in the pea rhizosphere, including Variovorax, and the archaeon Methanosarcina was also observed, reinforcing the role of C1 metabolism in rhizosphere colonisation.
Some taxa were selected by more than one plant, suggesting that they may be general rhizosphere colonisers. Among prokaryotes, only the Verrucomicrobiaceae were more abundant in all rhizospheres compared with bulk soil. There are few cultured representatives of this poorly understood group. Fungi, particularly Ascomycota, were selected by oat and strongly selected by pea, whereas fungi closely related to those that form lichens (Chaetothyriomycetidae), bacterivorous protozoa (Cercomonadida and Kinetoplastida) and nematodes (Criconematoidea) were selected by all plants. No eukaryotic taxa were statistically less abundant in a rhizosphere sample compared with bulk soil.
The production of antimicrobial compounds by plants and competition or inability to use plant-derived carbon sources inevitably leads to some taxa being depleted in the rhizosphere relative to soil. Sphingomondales and potentially plant pathogenic Xanthomonadales were less abundant in the wheat rhizosphere, Actinomycetales and Rhizobiales were less abundant in the oat rhizosphere, whereas sulphate-reducing Desulphobacteriales, bacterivorous Bdellovibrionaceae and nitrite-oxidising Nitrospira were less abundant in the pea rhizosphere.
Photosynthetic Noscocales, including Anabaena, were depleted in all rhizospheres. As plants provide the major carbon flux into the rhizosphere, microbial autotrophs may lose their selective advantage. A range of taxa, including Alphaproteobacteria, Bacillaceae and Acidimicrobiales, were depleted in both oat and pea rhizospheres. One explanation could be that these bacteria were grazed upon by nematodes and protozoa, which showed a higher relative abundance in all rhizospheres, but particularly those of oat and pea compared with bulk soil. Introducing a protozoan predator into the Arabidopsis thaliana rhizosphere altered relative abundance of specific bacterial groups, indicating the feeding preferences of protozoa (Rosenberg et al., 2009).
Comparison of the wild-type and sad1 mutant oat rhizospheres
Rarefaction analysis showed eukaryotic diversity in the sad1 rhizosphere to be greater than that of the wild-type and other environments at both the phylum and genus level, whereas prokaryotic diversity was unaltered (Supplementary Figure 2). Prokaryotic community profiles of both oat genotypes were similar at the phylum and genus level (Figure 1), although there were differences in relative abundance of some bacteria. Cyanobacteria, Spirochaetes and unclassified Bacillales were more abundant in the sad1 rhizosphere, whereas Streptomycetaceae, Nitrospinaceae and Sinobacteriaceae were less abundant. At both the phylum and genus level, eukaryotic communities were distinct. The sad1 rhizosphere was enriched for two fungal taxa, Mucoromycotina and Pezizomycetes, but there was no significant difference in overall fungal community compared with the wild type (Figure 2). The non-fungal eukaryotic community, particularly Amoebozoa and Alveolata, was more strongly affected (Figure 2). A few eukaryotes were more abundant in the wild-type oat rhizosphere, including Neoptera, Criconematoidea and Heteromitidae.
Avenacins have broad-spectrum anti-fungal activity (Maizel et al., 1964; Carter et al., 1999) and plants defective in their production have compromised resistance to fungal pathogens (Osbourn et al., 1994; Papadopoulou et al., 1999). It is surprising therefore that there was little difference between the fungal community of the wild-type and the avenacin-deficient mutant. Avenacins’ anti-fungal activity is attributed to their ability to bind to sterols, forming a pore that disrupts the cell membrane (Armah et al., 1999). Sterols are almost exclusively eukaryotic (Desmond and Gribaldo, 2009), which may account for the shift in eukaryotic community between the two oat genotypes tested here. Perhaps, avenacins have higher affinity for sterols in Amoebozoa and Alveolata than for those in fungi. Additional biological activities of avenacins are unknown and determining them is beyond the scope of this study. However, it is worth noting that small changes in plant genotype can have complex and unpredictable effects on the rhizosphere microbiome.
Conclusion
Using metatranscriptomics, the active rhizosphere microbiomes of several plants were characterised. Arguably, the most important organisms in an environment are metabolically active and metatranscriptomic analysis of rRNA is an indicator of this community. A common problem with metatranscriptomic studies is the absence of biological replicates due to the cost of sequencing. In this study, we have used 3–5 biological replicates to overcome this problem. The variation between biological replicates was modest, thus validating this approach. However, in future the use of Illumina sequencing (Bentley et al., 2008) with sequence reads of 250–500 bp, now available with the MiSeq, will allow both greatly increased sequence depth and replication, as demonstrated in a recent amplicon-based study (Caporaso et al., 2012). This will be particularly useful for determining the differential expression of metabolic pathways in different rhizospheres and detection of rare microorganisms. Metatranscriptomic studies have focussed on enriching mRNA from oceans (Poretsky et al., 2009; Shi et al., 2009), human gut (Gosalbes et al., 2011), soil (Bailly et al., 2007) and other environments, identifying active metabolic pathways. The soil study was restricted to eukaryotes (Bailly et al., 2007), because of the ease of mRNA enrichment. Thus, the global composition and metabolism of the rhizosphere microbiome is poorly understood.
After only 4 weeks of growth, the microbiomes of three crop plants were different from each other and from bulk soil, with a profound change in the balance of prokaryotes and eukaryotes between plants. Differences in the field are likely to be greater, as crops are typically grown for several months. Oat and pea exerted strong selection on eukaryotes, whereas selection by wheat was much weaker. Oat and pea are used extensively as break-crops in crop rotation systems. A recent meta-analysis showed increased wheat yield after seasons of either oat or pea (Seymour et al., 2012). The effect of pea was largely attributed to improved soil nitrogen status, whereas oat reduced disease incidence. The large shifts in rhizosphere microbiota seen here for oat and pea may also contribute to their positive effects when used in crop rotation systems (Figure 3a).
No eukaryotic taxa were significantly less abundant in the rhizosphere compared with bulk soil, with all rhizosphere samples showing higher eukaryotic diversity than bulk soil. This enrichment of eukaryotes, particularly nematodes and protozoa, implies that more resources are available from either the plant or its microbiome. As protozoan predators of bacteria have feeding preferences (Rosenberg et al., 2009), the relative abundance of some bacterial groups enriched in the rhizospheres may be because they avoided predation rather than they were selected directly by the plant. This emphasises the complexity of interactions occurring in the rhizosphere, and reinforces the value of global analysis.
Insight into functional roles in environments can be provided by the presence of well-characterised taxonomic groups. Cereal (wheat and oat) rhizospheres were enriched for cellulose degraders, whereas a legume (pea) rhizosphere was enriched for H2 oxidisers. Different methylotrophs were enriched in rhizospheres, depending on whether the plant was a cereal or a legume. However, the presence of an organism with the potential to carry out a biochemical process is not evidence for the activity of the metabolic process itself. Functional metatranscriptomics (i.e. analysis of mRNA) would be required to determine this, and is currently underway.
Metatranscriptomic analysis was sensitive enough to detect differences due to a single mutation in host plant. The loss of avenacin production in a mutant oat had broad effects on the eukaryotic community, whereas prokaryotes were weakly affected. This result could be related to the presence of sterols in the membranes of eukaryotes but not, with few exceptions, prokaryotes (Desmond and Gribaldo, 2009). The diversity of eukaryotes in the sad1 rhizosphere was consistently greater than the wild-type and the other environments. These differences would likely have been missed by PCR-based analysis of bacteria or fungi because it was the non-fungal eukaryotic community that was most strongly influenced.
Metatranscriptomics is a powerful tool in microbial ecology and can provide an initial, comprehensive picture of community structure from an environment. If desired, targeted PCR can then be used to focus on important groups. Here, this global approach highlighted the complexity of the rhizosphere microbiome and revealed profound differences in community structure between plants, particularly at the kingdom level.
References
Alon S, Vigneault F, Eminaga S, Christodoulou DC, Seidman JG, Church GM et al (2011). Barcoding bias in high-throughput multiplex sequencing of miRNA. Genome Res 21: 1506–1511.
Armah CN, Mackie AR, Roy C, Price K, Osbourn AE, Bowyer P et al (1999). The membrane-permeabilizing effect of avenacin A-1 involves the reorganization of bilayer cholesterol. Biophys J 76: 281–290.
Bailly J, Fraissinet-Tachet L, Verner MC, Debaud JC, Lemaire M, Wesolowski-Louvel M et al (2007). Soil eukaryotic functional diversity, a metatranscriptomic approach. ISME J 1: 632–642.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG et al (2008). Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59.
Bloemberg GV, Lugtenberg BJJ . (2001). Molecular basis of plant growth promotion and biocontrol by rhizobacteria. Curr Opin Plant Biol 4: 343–350.
Bonfante P . (2010) Plant–Fungal Interactions in Mycorrhizas. Wiley: New York, NY, USA.
Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, Assenza F et al (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488: 91–95.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N et al (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6: 1621–1624.
Carter JP, Spink J, Cannon PF, Daniels MJ, Osbourn AE . (1999). Isolation, characterization, and avenacin sensitivity of a diverse collection of cereal-root-colonizing fungi. Appl Environ Microbiol 65: 3364–3372.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Delmotte N, Knief C, Chaffron S, Innerebner G, Roschitzki B, Schlapbach R et al (2009). Community proteogenomics reveals insights into the physiology of phyllosphere bacteria. Proc Natl Acad Sci USA 106: 16428–16433.
Dennis PG, Miller AJ, Hirsch PR . (2010). Are root exudates more important than other sources of rhizodeposits in structuring rhizosphere bacterial communities? FEMS Microbiol Ecol 72: 313–327.
Desmond E, Gribaldo S . (2009). Phylogenomics of sterol synthesis: insights into the origin, evolution, and diversity of a key eukaryotic feature. Genome Biol Evol 1: 364–381.
Dong Z, Wu L, Kettlewell B, Caldwell CD, Layzell DB . (2003). Hydrogen fertilization of soils—is this a benefit of legumes in rotation? Plant Cell Environ 26: 1875–1879.
Dray S, Dufour AB . (2007). The ade4 package: Implementing the duality diagram for ecologists. J Statist Softw 22: 1–20.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Feil H, Feil WS, Chain P, Larimer F, DiBartolo G, Copeland A et al (2005). Comparison of the complete genome sequences of Pseudomonas syringae pv. syringae B728a and pv. tomato DC3000. Proc Natl Acad Sci USA 102: 11064–11069.
Galbally IE, Kirstine W . (2002). The production of methanol by flowering plants and the global cycle of methanol. J Atmos Chem 43: 195–229.
Gomez-Alvarez V, Teal TK, Schmidt TM . (2009). Systematic artifacts in metagenomes from complex microbial communities. ISME J 3: 1314–1317.
Gosalbes MJ, Durban A, Pignatelli M, Abellan JJ, Jimenez-Hernandez N, Perez-Cobas AE et al (2011). Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS One 6: 9.
Han JI, Choi HK, Lee SW, Orwin PM, Kim J, Laroe SL et al (2011). Complete genome sequence of the metabolically versatile plant growth-promoting endophyte variovorax paradoxus S110. J Bacteriol 193: 1183–1190.
Handelsman J . (2004). Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68: 669–685.
Haralampidis K, Bryan G, Qi X, Papadopoulou K, Bakht S, Melton R et al (2001). A new class of oxidosqualene cyclases directs synthesis of antimicrobial phytoprotectants in monocots. Proc Natl Acad Sci USA 98: 13431–13436.
Hong SH, Bunge J, Leslin C, Jeon S, Epstein SS . (2009). Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J 3: 1365–1373.
Hunt S, Layzell DB . (1993). Gas-exchange of legume nodules and the regulation of nitrogenase activity. Annu Rev Plant Physiol Plant Mol Biol 44: 483–511.
Huson DH, Auch AF, Qi J, Schuster SC . (2007). MEGAN analysis of metagenomic data. Genome Res 17: 377–386.
Inceoglu O, Abu Al-Soud W, Salles JF, Semenov AV, van Elsas JD . (2011). Comparative analysis of bacterial communities in a potato field as determined by pyrosequencing. PLoS One 6: 11.
Janssen PH . (2006). Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72: 1719–1728.
Kalyuhznaya MG, Martens-Habbena W, Wang TS, Hackett M, Stolyar SM, Stahl DA et al (2009). Methylophilaceae link methanol oxidation to denitrification in freshwater lake sediment as suggested by stable isotope probing and pure culture analysis. Environ Microbiol Rep 1: 385–392.
Knief C, Delmotte N, Chaffron S, Stark M, Innerebner G, Wassmann R et al (2012). Metaproteogenomic analysis of microbial communities in the phyllosphere and rhizosphere of rice. ISME J 6: 1378–1390.
Lu YH, Rosencrantz D, Liesack W, Conrad R . (2006). Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ Microbiol 8: 1351–1360.
Lugtenberg B, Kamilova F . (2009). Plant-growth-promoting rhizobacteria. Annu Rev Microbiol (Annu Rev: Palo Alto) 63: 541–556.
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S et al (2012). Defining the core Arabidopsis thaliana root microbiome. Nature 488: 86–90.
Maimaiti J, Zhang Y, Yang J, Cen YP, Layzell DB, Peoples M et al (2007). Isolation and characterization of hydrogen-oxidizing bacteria induced following exposure of soil to hydrogen gas and their impact on plant growth. Environ Microbiol 9: 435–444.
Maizel JV, Mitchell HK, Burkhardt HJ . (1964). Avenacin antimicrobial substance isolated from avena sativa.1. Isolation+antimicrobial activity. Biochemistry 3: 424–426.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA et al (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380.
Mendes R, Kruijt M, de Bruijn I, Dekkers E, van der Voort M, Schneider JHM et al (2011). Deciphering the rhizosphere microbiome for disease-suppressive bacteria. Science 332: 1097–1100.
Osbourn AE, Clarke BR, Lunness P, Scott PR, Daniels MJ . (1994). An oat species lacking avenacin is susceptible to infection by Gaeumannomyces-Graminis var tritici. Physiol Mol Plant Pathol 45: 457–467.
Ottesen EA, Marin R, Preston CM, Young CR, Ryan JP, Scholin CA et al (2011). Metatranscriptomic analysis of autonomously collected and preserved marine bacterioplankton. ISME J 5: 1881–1895.
Papadopoulou K, Melton RE, Leggett M, Daniels MJ, Osbourn AE . (1999). Compromised disease resistance in saponin-deficient plants. Proc Natl Acad Sci USA 96: 12923–12928.
Paulsen IT, Press CM, Ravel J, Kobayashi DY, Myers GSA, Mavrodi DV et al (2005). Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23: 873–878.
Philippot L, Hallin S, Borjesson G, Baggs EM . (2009). Biochemical cycling in the rhizosphere having an impact on global change. Plant Soil 321: 61–81.
Pinto AJ, Raskin L . (2012). PCR biases distort bacterial and archaeal community structure in pyrosequencing datasets. PLoS One 7: 16.
Poretsky RS, Hewson I, Sun SL, Allen AE, Zehr JP, Moran MA . (2009). Comparative day/night metatranscriptomic analysis of microbial communities in the North Pacific subtropical gyre. Environ Microbiol 11: 1358–1375.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig WG, Peplies J et al (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Ramachandran VK, East AK, Ramakrishnan K, Downie JA, Poole PS . (2011). Adaptation of Rhizobium leguminosarum to pea, alfalfa and sugar beet rhizospheres investigated by comparative transcriptomics. Genome Biol 12: R106.
Ransom-Jones E, Jones DL, McCarthy AJ, McDonald JE . (2012). The fibrobacteres: an important phylum of cellulose-degrading bacteria. Microbial Ecol 63: 267–281.
Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD et al (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Rosenberg K, Bertaux J, Krome K, Hartmann A, Scheu S, Bonkowski M . (2009). Soil amoebae rapidly change bacterial community composition in the rhizosphere of arabidopsis thaliana. ISME J 3: 675–684.
Ryan RP, Monchy S, Cardinale M, Taghavi S, Crossman L, Avison MB et al (2009). The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat Rev Microbiol 7: 514–525.
Seymour M, Kirkegaard JA, Peoples MB, White PF, French RJ . (2012). Break-crop benefits to wheat in Western Australia—insights from over three decades of research. Crop Past Sci 63: 1–16.
Shi YM, Tyson GW, DeLong EF . (2009). Metatranscriptomics reveals unique microbial small RNAs in the ocean’s water column. Nature 459: 266–U154.
Shi YM, Tyson GW, Eppley JM, DeLong EF . (2011). Integrated metatranscriptomic and metagenomic analyses of stratified microbial assemblages in the open ocean. ISME J 5: 999–1013.
Stewart FJ, Ottesen EA, DeLong EF . (2010). Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J 4: 896–907.
Stracke S, Kistner C, Yoshida S, Mulder L, Sato S, Kaneko T et al (2002). A plant receptor-like kinase required for both bacterial and fungal symbiosis. Nature 417: 959–962.
Teixeira L, Peixoto RS, Cury JC, Sul WJ, Pellizari VH, Tiedje J et al (2010). Bacterial diversity in rhizosphere soil from Antarctic vascular plants of Admiralty Bay, maritime Antarctica. ISME J 4: 989–1001.
Tett AJ, Turner TR, Poole PS . (2012) Genomics and the Rhizosphere. Wiley: New York, NY, USA.
Urich T, Lanzen A, Qi J, Huson DH, Schleper C, Schuster SC . (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3: e2527.
Yashiro E, Spear RN, McManus PS . (2011). Culture-dependent and culture-independent assessment of bacteria in the apple phyllosphere. J Appl Microbiol 110: 1284–1296.
Yi H, Cho YJ, Won S, Lee JE, Yu HJ, Kim S et al (2011). Duplex-specific nuclease efficiently removes rRNA for prokaryotic RNA-seq. Nucleic Acids Res 39: 9.
Young JPW, Crossman LC, Johnston AWB, Thomson NR, Ghazoui ZF, Hull KH et al (2006). The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol 7: 20.
Yu L, Nicolaisen M, Larsen J, Ravnskov S . (2012). Molecular characterization of root-associated fungal communities in relation to health status of Pisum sativum using barcoded pyrosequencing. Plant Soil 357: 395–405.
Acknowledgements
We thank the Earth and Life Systems alliance of the Norwich Research Park for funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Turner, T., Ramakrishnan, K., Walshaw, J. et al. Comparative metatranscriptomics reveals kingdom level changes in the rhizosphere microbiome of plants. ISME J 7, 2248–2258 (2013). https://doi.org/10.1038/ismej.2013.119
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2013.119
Keywords
This article is cited by
-
Soil microbial ecology through the lens of metatranscriptomics
Soil Ecology Letters (2024)
-
Study on the soil microbial community structure of the Rhizosphere of Camellia sinensis L. in Anping Village, Kaiyang County, Guizhou Province
Annals of Microbiology (2023)
-
16S metabarcoding, total soil DNA content, and functional bacterial genes quantification to characterize soils under long-term organic and conventional farming systems
Chemical and Biological Technologies in Agriculture (2023)
-
Genome-resolved metatranscriptomics reveals conserved root colonization determinants in a synthetic microbiota
Nature Communications (2023)
-
Changes in soil microbial diversity and community composition across bahiagrass and rhizoma peanut pastures
Biology and Fertility of Soils (2023)

{kind=link}
{kind=link}