
Abstract
Soils of coniferous forest ecosystems are important for the global carbon cycle, and the identification of active microbial decomposers is essential for understanding organic matter transformation in these ecosystems. By the independent analysis of DNA and RNA, whole communities of bacteria and fungi and its active members were compared in topsoil of a Picea abies forest during a period of organic matter decomposition. Fungi quantitatively dominate the microbial community in the litter horizon, while the organic horizon shows comparable amount of fungal and bacterial biomasses. Active microbial populations obtained by RNA analysis exhibit similar diversity as DNA-derived populations, but significantly differ in the composition of microbial taxa. Several highly active taxa, especially fungal ones, show low abundance or even absence in the DNA pool. Bacteria and especially fungi are often distinctly associated with a particular soil horizon. Fungal communities are less even than bacterial ones and show higher relative abundances of dominant species. While dominant bacterial species are distributed across the studied ecosystem, distribution of dominant fungi is often spatially restricted as they are only recovered at some locations. The sequences of cbhI gene encoding for cellobiohydrolase (exocellulase), an essential enzyme for cellulose decomposition, were compared in soil metagenome and metatranscriptome and assigned to their producers. Litter horizon exhibits higher diversity and higher proportion of expressed sequences than organic horizon. Cellulose decomposition is mediated by highly diverse fungal populations largely distinct between soil horizons. The results indicate that low-abundance species make an important contribution to decomposition processes in soils.
Similar content being viewed by others


Introduction
Most terrestrial ecosystem functions occur in the soil, which possesses the greatest amount of biodiversity on Earth. Yet, the understanding of how ecosystem functions are influenced by soil biodiversity is far behind our understanding of how aboveground organisms contribute to these functions (Bowker et al., 2010). Soil microorganisms represent a considerable fraction of the living biomass on Earth, with 103–104 kg of microbial biomass per hectare of surface soils (Fierer et al., 2007). In addition, microbial community composition is now recognised as an important determinant of ecosystem process rates (Reed and Martiny 2007; Strickland et al., 2009). Understanding the structure and function of soil microbial communities is thus central to predicting how ecosystems will respond to future environmental conditions.
While several recent studies have used deep sequencing approaches to assess the diversity of soil bacterial components (Roesch et al., 2007; Lauber et al., 2009), the number of such studies addressing fungal diversity is still limited. This is true despite the fact that fungi comprise a large proportion of soil microbial biomass and have a dominant role in decomposition and nutrient cycling in soil (Bailey et al., 2002; Buée et al., 2009). Only a minor fraction of the estimated 1.5 million fungal species worldwide have been described (Hawksworth, 2001), and the ecological roles of most fungal taxa are poorly understood since the complexity of fungal communities has so far limited our ability to estimate diversity and distinguish individual taxa (McGuire and Treseder, 2010). In order to understand the soil ecosystem processes, it is essential to address the fungal and bacterial community at the same time. In addition, the ecology of total microbial communities is so far largely derived from the studies on DNA and no information is available on the relationships between the diversity of this total community and community of active microbes assessed, for example, by targeting the RNA molecules (Anderson and Parkin, 2007; Urich et al., 2008).
From the global viewpoint, the understanding of fungal and bacterial diversity is highly important in the biomes of coniferous forests, where fungi quantitatively dominate bacteria in decomposing litter material, while the importance of bacteria increases with soil depth (Bååth and Anderson 2003). Coniferous forest ecosystems have a prominent role in the global carbon cycle (Myneni et al., 2001), and knowledge of microbially mediated soil functions is thus required for estimating global C fluxes and their potential future changes. Forests dominated by spruce (Picea spp.) constitute large ecosystems in boreal forest biomes and are also widely distributed in higher altitude forests and plantation forests in the northern temperate zone.
It was recently proposed that the analyses of soil microbial community composition should be based on direct analysis of total RNA to avoid PCR bias (Urich et al., 2008). While this approach may be feasible for bacterial community analyses, the sequence information contained in fungal rRNA molecules is insufficient for species discrimination; thus, internal transcribed spacer (ITS) regions of the rRNA are used instead. Because there are 103–104 times fewer fungal ITS sequences than bacterial 16S rRNA gene sequences in soil (Figure 1), amplification of fungal ITS is inevitable to achieve reasonable sampling depth. Here, we combined the analysis of DNA-derived bacterial 16S rRNA gene sequences representing all bacteria present and the RNA-derived sequences representing the content of bacterial ribosomes reflecting thus the active part of the total community. The analysis of fungal ITS1 and ITS2 sequences offers a unique opportunity to target the precursor rRNA molecules with fast turnover, thus identifying these species synthesising ribosomes at a given moment and thus likely metabolically active (Anderson and Parkin, 2007). The comparison of the DNA and RNA communities can also help to answer the question how well are the metabolically active microbial taxa represented in the common studies using the sequencing of soil DNA.

Properties of Picea abies forest soil, abundance of microorganisms and activity of extracellular enzymes involved in organic matter decomposition in the L and H horizons. The data represent mean values and s.d. from four studied sites.
The aim of this work was to demonstrate how the DNA and RNA communities differ and what part of the total community is metabolically active at a given moment. The study was performed at the beginning of winter under freshly fallen snow to target the period when decomposition processes in soil prevail. In a mountainous Picea abies forest in central Europe where mycorrhizal fungi have a major role, the winter period without photosynthate flow is expected to show increased activity of decomposer fungal species. Litter and the organic horizons were studied separately because fungal and bacterial communities were previously found to differ between these horizons due to the differences in nutrient availability and the presence of root-associated microorganisms (O’Brien et al., 2005; Lindahl et al., 2007; Šnajdr et al., 2008).
We expected that during the decomposition period, decomposer microorganisms will be transcriptionally active and a large proportion of decomposition-related genes will be expressed by dominant taxa of microbial decomposers. In order to specifically target an important decomposition process, the gene and transcriptome pools of the fungal cellobiohydrolase (exocellulase) gene cbhI sequences were compared, as its gene product catalyses the rate-limiting step in the decomposition of cellulose, the most abundant biopolymer in the ecosystem (Baldrian and Valášková, 2008). The cbhI gene occurs in both Ascomycota and Basidiomycota, and it is also common in the genomes of saprotrophic fungi (Edwards et al., 2008; Ohm et al., 2010). While some recent studies showed that expression of eukaryotic decomposition-related genes in soils can be analysed (Luis et al., 2005; Kellner and Vandenbol, 2010), only the DNA/RNA approach can answer the questions on the diversity of decomposer communities and the proportion of expressed genes. Since cellulose is present in both the litter and soil organic horizon (Šnajdr et al., 2011), the same cellulose-decomposing microorganisms can be present and potentially active in both horizons. However, the higher amount of cellulose in litter likely supports higher diversity of cellulose decomposers.
Materials and methods
Study site, sample collection and soil analysis
Study area was located in the highest altitudes (1170–1200 m) of the Bohemian Forest mountain range (Central Europe) and was covered by an unmanaged spruce (P. abies) forest (49°02.64 N, 13°37.01 E). Sampling was performed in late October 2009 under freshly fallen snow (8–12 cm, 3 days after the snowfall) at −5 °C. At four sites, located 250 m from each other, six topsoil samples located around the circumference of a 4-m-diameter circle were collected. Litter horizon (L) and organic (humic) horizon (H) material were separately pooled. After removal of roots, L material was cut into 0.5 cm pieces and mixed; H material was passed through a 5-mm sterile mesh and mixed. Aliquots for nucleic acids extraction were immediately frozen and stored in liquid nitrogen. Samples for phospholipid fatty acid and ergosterol analysis were frozen and stored at −45 °C until analysis. Enzyme assays were performed within 48 h in samples kept at 4 °C in soil homogenates (Štursová and Baldrian, 2011). Dry mass content was measured after drying at 85 °C, organic matter content after burning at 650 °C and pH was measured in distilled water (1:10). Soil C and N content was measured using an elemental analyser.
Quantification of microbial biomass
Phospholipid fatty acid was extracted by chloroform–methanol–phosphate buffer, subjected to alkaline methanolysis and free methyl esters were analysed by GC-MS (Šnajdr et al., 2008). Fungal biomass was quantified based on 18:2ω6,9 content, and bacterial biomass as the sum of bacteria-specific phospholipid fatty acid (Bååth and Anderson, 2003). Total ergosterol was extracted with 10% KOH in methanol and analysed by HPLC (Šnajdr et al., 2008). Partial bacterial and fungal rDNAs were quantified by qPCR using 1108f and 1132r primers for bacteria (Wilmotte et al., 1993; Amann et al., 1995) and ITS1/qITS2* primers for fungi (White et al., 1990; Šnajdr et al., 2011).
Nucleic acid extraction and reverse transcription
RNA and DNA were co-extracted using the RNA PowerSoil Total RNA Isolation Kit and DNA Elution Accessory Kit (MoBio Laboratories, Carlsbad, CA, USA) combined with the OneStep PCR Inhibitor Removal Kit (Zymo Research, Irvine, CA, USA). Three soil aliquots (3 × 3 g of material) were extracted per sample. Extracted RNA was treated with DNase I and 1 μg was reverse transcribed using M-MLV Reverse Transcriptase (Invitrogen, Carlsbad, CA, USA) and random hexamer primers. Samples were designated as LD=litter DNA, LR=litter cDNA, HD=humic horizon DNA and HR=humic horizon cDNA.
Tag-encoded amplicon pyrosequencing and sequence analysis
The eubacterial primers eub530F/eub1100aR (modified from Dowd et al., 2008) were used to amplify the V4–V6 region of bacterial 16S rDNA and the fungi-specific primers ITS1/ITS4 (White et al., 1990) were used to amplify the ITS1, 5.8S rDNA and ITS2 regions of fungal rDNA. Primers cbhIF and cbhIR (Edwards et al., 2008) were used to amplify a partial sequence of fungal cellobiohydrolase I. Primers for tag-encoded 454-Titanium pyrosequencing contained in addition sample tags separated from primers by spacers and Titanium A or B adaptors (Roche, Basel, Switzerland). Spacer sequences were designed to contain a trinucleotide, absent in all GenBank sequences at this position to avoid preferential amplification of some targets (Parameswaran et al., 2007). Primer pairs were designed using OligoCalc (http://www.basic.northwestern.edu/biotools/oligocalc.html) and tested by cloning/sequencing. Tags and spacer sequences of all composite primers used for tag-encoded amplicon pyrosequencing in this study can be found in Supplementary Information.
PCR amplifications were performed in two steps. In the first step, each of three independent 50 μl reactions per DNA/cDNA sample contained 5 μl of 10 × polymerase buffer, 3 μl of 10 mg ml−1 bovine serum albumin, 2 μl of each primer (0.01 mM), 1 μl of PCR Nucleotide Mix (10 mM), 1.5 μl polymerase (2 U μl−1; Pfu DNA polymerase:DyNAZyme II DNA polymerase, 1:24) and 2 μl of template DNA. Cycling conditions were 94 °C for 5 min; 35 cycles of 94 °C for 1 min, 62 °C for 50 s, 72 °C for 30 s, followed by 72 °C for 10 min for primers eub530F/eub1100aR; 94 °C for 5 min; 35 cycles of 94 °C for 1 min, 55 °C for 1 min, 72 °C for 1 min, followed by 72 °C for 10 min for primers ITS1/ITS4; 94 °C for 3 min; 40 cycles of 94 °C for 30 s, 51 °C for 45 s, 72 °C for 1 min 30 s, followed by 72 °C for 15 min for primers cbhIF/cbhIR. Pooled PCR products were purified using the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). In all, 100 ng DNA was used as template in the second PCR performed under the same conditions except that fusion primers were used and cycle number was 10. PCR products were separated by electrophoresis and gel purified using the Wizard SV Gel and PCR Clean-Up System. DNA was quantified using ND1000 (NanoDrop, Wilmington, DE, USA), an equimolar mix of PCR products from all samples was made for each primer pair and the pooled products were mixed in a molar ratio of 12:4:1 (bacterial:fungal:cbhI amplicons). The mixture was subjected to sequencing on a GS FLX Titanium platform (Roche).
The pyrosequencing resulted in 329 820 reads of sufficient quality and a length >200 bases. Pyrosequencing noise reduction was performed using the Denoiser 0.851 (Reeder and Knight, 2010) and chimeric sequences were detected using UCHIME (Edgar, 2010) and deleted. In fungal community analyses, sequences >380 bases were used that contained the ITS1 region, 5.8S rDNA and a significant part of the ITS2 region. These sequences were truncated to 380 bases, clustered using CD-HIT (Li and Godzik, 2006) at 97% similarity (O’Brien et al., 2005) to yield Operational Taxonomic Units (OTUs) and consensus sequences were constructed for all OTUs. PlutoF pipeline (Tedersoo et al., 2010) was used to generate best species hits. In bacterial analysis, sequences of 350 bases were clustered at a 97% similarity and Ribosomal Database Project (Cole et al., 2009) as well as BLASTn hits against GenBank were used to generate best hits (Altschul et al., 1997). DNA/RNA ratio was calculated as sum of sequences derived from DNA divided by the sum of all sequences, and the L/H ratio was calculated similarly. Clusters of cbhI sequences were constructed using 400-base sequences at 96% similarity. Intron positions were recorded and introns removed from the DNA-derived sequences and DNA and cDNA-derived clusters were merged. For identification, cbhI sequences were retrieved from GenBank and also obtained by the analysis of isolates or cultured strains from the studied ecosystem by cloning/sequencing. Nucleotide sequences of OTUs with abundances over 0.3% were translated into amino-acid sequences in Bionumerics 7.0 (Applied Maths, Sint-Martens-Latem, Belgium).
Rarefaction and diversity analyses on OTUs/clusters were performed at 8500 bacterial, 1000 fungal or 350 cbhI sequences per sample, to eliminate the effect of sampling effort and used for clustering as described above. Richness and diversity indices were calculated using EstimateS 8.00 (http://viceroy.eeb.uconn.edu/estimates) and quality of Chao1 estimates was evaluated according to Kemp and Aller (2004).
One-way analysis of variance (ANOVA) with the Fisher's LSD post hoc test was used to analyse the statistical differences among treatments. To analyse the differences in bacterial and fungal communities and the cbhI sequences, principal component analysis was run with abundance data of all OTUs or clusters with >0.3% abundance. PC1 and PC2 loads were subjected to ANOVA with the Fisher's LSD post hoc test. Differences at P<0.05 were regarded as statistically significant.
Results and discussion
Microbial communities in P. abies topsoil are diverse and vertically stratified
The topsoil of the P. abies forest was strongly acidic (pH 3.7–3.8) and consisted of a 1–4-cm-thick litter horizon (L) and a 2–4-cm-thick organic (humic) horizon (H). The horizons were significantly different with respect to organic matter and C and N contents, and decreasing nutrient availability was reflected by a decrease in both bacterial and fungal biomass contents with depth. The results of qPCR showed a decrease of fungal-to-bacterial rDNA copy number ratio from 1.11 in the L horizon to 0.30 in the H horizon. The cDNA contained 105–106 copies of the bacterial 16S rRNA gene per nanogram cDNA, but only 102–104 copies per nanogram cDNA of fungal ITS region (Figure 1). More rapid organic matter transformation occurred in the L horizon than in the H horizon, as documented by higher activities of several extracellular enzymes, especially those hydrolysing glucans (α- and β-glucosidase and cellobiohydrolase; Figure 1).
Bacterial communities analysed at 8500 sequences per sample showed about 1500 OTUs per sample in the L and H horizon-derived DNA (LD and HD) and in the L-derived RNA (LR) samples, about 1200 OTUs were identified in the H-derived RNA (HR). Also, the Chao1 estimator predictions were lower for HR. The RNA-derived communities were less diverse and less even than the DNA-derived communities, particularly in the H horizon (Supplementary Table 1). Principal component analysis followed by ANOVA showed significant differences in community composition among LD, HD, LR and HR (P<0.00001 for differences among L and H as well as among DNA/RNA).
In the DNA community, Steroidobacter (OTU4) was the most abundant in all L samples. In the H horizon, Gp1 Acidobacterium (OTU1) was the most abundant at three sites and Gp2 Acidobacterium (OTU5) at one site (Supplementary Table 2; Figure 1). Members of 505 genera were found in the entire pooled community, with most of the sequences belonging to Gp1, Gp3 and Gp2 Acidobacteria (on average 17.2%, 11.2% and 8.4%, respectively), the Actinobacteria Actinoallomurus (7.1%), Conexibacter (1.3%) and Iamia (1.1%), and Proteobacteria Steroidobacter (5.4%), Rhodoplanes (3.3%), Phenylobacterium (2.1%), Desulfomonile (1.7%) and Burkholderia (1.5%; Supplementary Table 2). Of the most abundant OTUs, 33 (Chondromyces), 53 and 20 (Phenylobacterium) and 39 (Caulobacteraceae) were identified as highly enriched in the RNA-derived community and several taxa showed preferential association with either the L or the H horizon (Figure 2; Supplementary Table 2).

Distribution of major bacterial and fungal OTUs and cbhI clusters from Picea abies forest topsoil between the L and H horizons and between DNA and RNA. The data represent mean values from four sampling sites. Symbol areas correspond to relative abundance in the combined set of DNA and RNA sequences from both horizons. (a) Bacteria, identifications: 0=Actinoallomurus; 1=Gp1 Acidobacterium; 2=Rhodoplanes; 3=Rhodospirillales; 4=Steroidobacter; 5=Gp2 Acidobacterium; 6=Gp1 Acidobacterium; 7=Rhizobiales; 8=Gp2 Acidobacterium; 10=Gp1 Acidobacterium; 11=Frankineae; 12=Gp3 Acidobacterium; 13=Afipia; 14=Gp2 Acidobacterium; 15=Burkholderia; 16=Actinomycetales; 19=Gp3 Acidobacterium; 20=Phenylobacterium; 21=Desulfomonile; 22=Gp3 Acidobacterium; 23=Gp3 Acidobacterium; 24=Ferrithrix; 25=Acetobacteraceae; 26=Rhizobiales; 27=Gp3 Acidobacterium; 28=Gp1 Acidobacterium; 30=Gp3 Acidobacterium; 31=Acidisphaera; 32=Actinoallomurus; 33=Gp1 Acidobacterium; 34=Sporomusa; 35=Chondromyces; 36=Acetobacteraceae; 37=Steroidobacter; 38=Chitinophagaceae; 39=Caulobacteraceae; 40=Rhizobiales; 41=Mycobacterium; 48=Gp1 Acidobacterium; 59=Phenylobacterium. (b) Fungi, OTU identifications: 1=Ascomycete; 2=Tylospora fibrillosa; 3=Piloderma; 4=Piloderma; 5=Ascomycete; 6=Tylospora asterophora; 7=Cenococcum geophilum; 8=Verrucaria; 9=Hygrophorus olivaceoalbus; 10=Russula cyanoxantha; 11=Cortinarius biformis; 13=Lecanora; 14=Tylospora fibrillosa; 18=Cladophialophora minutissima; 20=Auriculoscypha; 22=Inocybe; 23=Ascomycete; 24=Basidiomycete; 25=Ascomycete; 27=Cryptococcus podzolicus; 28=Mycocentrospora acerina; 29=Ascomycete; 30=Ascomycete; 32=Meliniomyces vraolstadiae; 33=Amanita spissa; 44=Phellopilus; 45=Chytridiomycete; 47=Alternaria alternata; 49=Cenococcum geophilum; 50=Cortinarius gentilis; 52=Piloderma; 55=Neofusicoccum; 60=Russula cyanoxantha; 62=Trichosporon porosum; 67=Pseudotomentella; 69=Russula cyanoxantha; 70=Mycoarthris; 88=Elaphocordyceps; 94=Tomentella sublilacina. (c) cbhI, clusters with sequence similarities to genes of known cbhI producers: 0=Mycena; 1=Xylariales spp.; 16=Phacidium; 18=Mycena; 25=Phacidium; 28=Xylariales spp.; 42=Phialophora; 53=Xylariales spp.; 72=Phialophora; 75=Ceuthospora and 84=Phialophora sp.
Bacterial sequences belonged to 21 phyla, but only 8 were recorded with abundances over 0.1%. In both horizons, Proteobacteria, Acidobacteria and Actinobacteria were dominant, comprising 80–90% of all sequences; this dominance was even stronger in the RNA (Figure 3). In the L horizon, the RNA community was enriched in Acidobacteria and Firmicutes, while most of the minor phyla were less represented. In the H horizon, Actinobacteria were more abundant in the RNA community (Supplementary Table 2; Supplementary Figure 1).

Phylogenetic assignment of bacterial, fungal and cbhI sequences from Picea abies forest topsoil. The data represent mean values from four study sites. (a) Bacteria, (b) fungi and (c) cbhI sequences.
Bacterial abundance and diversity have been reported to decrease with decreasing soil pH (Lauber et al., 2009; Rousk et al., 2010). Despite this, a highly diverse bacterial community was found in our strongly acidic soil. Compared with other soils with pH <4, in which a high degree of dominance by Acidobacteria, around 63%, was previously reported (Lauber et al., 2009), the phyla Proteobacteria, Actinobacteria, Firmicutes and Verrucomicrobia were more represented in the P. abies forest. The bacterial community in the soil of the study area is specific in several aspects. The genus Chitinophaga, which was abundant in a previous study that compared different soils (Fulthorpe et al., 2008), was found at a frequency of only 0.1% in this study; the genera Actinoallomurus and Steroidobacter, ranked among the five most abundant genera in the P. abies forest, were not recovered in the previous study.
Deep sequencing analyses of bacterial communities associated with litter have not previously been reported. Here, we show that the litter horizon exhibited higher phylogenetic diversity and a reduced proportion of Acidobacteria in the total community. In the most abundant phylum, Acidobacteria, the members of Gp1, Gp2 and Gp3 detected were also reported to occur in other low pH soils (Jones et al., 2009). Genome sequencing of Gp1 and Gp3 Acidobacteria and isolate culturing showed that these bacteria are able to decompose a variety of polysaccharides, including cellulose, xylan and chitin, and thus may be involved in decomposition (Ward et al., 2009). Indeed, in our study, the Gp3 Acidobacteria were preferentially detected in the RNA, which indicates their activity during the decomposition period. The ecology of the other most abundant bacterial genera is unclear. Members of the genus Actinoallomurus (formerly belonging to Actinomadura) were repeatedly isolated from soils or litter, and some species from this genus are root endophytes. The genus Phenylobacterium contains bacteria from upper aerobic soil horizons capable of phenolic compound degradation.
Fungal communities sampled at 1000 randomly selected sequences per sample had between 150 and 220 OTUs with no significant differences between the DNA and RNA samples. The Chao1 estimates predicted higher diversity in LD and HD than in LR and HR (Supplementary Table 1). Principal component analysis followed by ANOVA showed significant differences in community composition among LD, HD, LR and HR (P<0.0005 for differences among L and H and P<0.044 for DNA/RNA).
In the DNA community, Piloderma sp. (OTU3) was dominant at two sites and Tylospora fibrillosa (OTU2) and Cortinarius biformis (OTU11) were each dominant at one site in the L horizon. In the H horizon, each of the sites was dominated by a different OTU (Tylospora fibrillosa, OTU2; Tylospora asterophora, OTU6; Russula cyanoxantha, OTU10; and Piloderma sp., OTU4). OTUs with the closest similarity to 422 different genera were recorded, the most abundant being Tylospora (14.8% of all sequences), Piloderma (12.8%), Russula (4.4%), Cenococcum (4.2%), Cortinarius (3.9%), Hygrophorus (2.9%), Cladophialophora (2.4%), Amanita (1.8%), Cadophora (1.7%), Mortierella (1.6%) and Verrucaria (1.6%; Supplementary Table 3). The distribution of the abundant OTUs among the L and H horizons and the DNA and RNA communities shows a strict confinement of many OTUs to either the L or the H horizon (Figure 2; Supplementary Table 3). Several of the most abundant OTUs were highly enriched in the RNA community (Supplementary Table 3).
Fungal sequences belonged mainly to Dikarya (53.5% Basidiomycota and 41.1% Ascomycota). Glomeromycota were represented by 2.24% of the sequences, Mucoromycotina by 1.77%, and Chytridiomycota by 0.73 (Supplementary Table 3). For several groups of phylogenetically related OTUs abundant in the ecosystem, no close sequence of an isolated strain was available. Some of these, including also the putative members of basal fungal lineages, exhibited high abundance in the RNA samples (Supplementary Figure 2). Members of the orders Atheliales, Agaricales, Helotiales, Chaetothyriales and Russulales were most abundant in the soil DNA; several minor orders, including Botryosphaeriales, Lecanorales and Eurotiales in the L horizon and Tremellales and Capnodiales in the H horizon, were infrequent in the DNA communities but highly abundant among the RNA sequences (Figure 3).
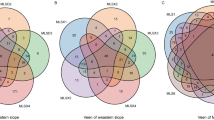
The composition of fungal communities has been previously shown to differ substantially between litter and organic horizons, while deeper soil horizons showed greater similarity (O’Brien et al., 2005; Lindahl et al., 2007). In several forest types, this is due to the higher abundance of saprotrophic fungi in litter and the dominance of ectomycorrhizal species in deeper soil (Lindahl et al., 2007; Edwards and Zak, 2010). Although our results showed that ectomycorrhizal fungi were highly dominant in both horizons (most likely due to the shallow rooting of P. abies), we also confirmed profound differences between the two horizons; 42% of abundant species were only recovered from either the L or the H horizon (Figure 4). The most abundant genera of ectomycorrhizal fungi found in this study, Piloderma and Tylospora spp., are also the most abundant in the boreal P. abies forests in Finland and Sweden (Rosling et al., 2003; Korkama et al., 2006; Wallander et al., 2010).

Distribution of bacterial and fungal OTUs and cbhI clusters from Picea abies forest soil among the L and H horizons and among DNA and RNA, in percents. Based on the data for OTUs/clusters with abundance >0.1%. The y axis represents relative share of transcripts in the L horizon or in the DNA. The values 0.0 and 1.0 represent OTUs/clusters present only in either the H or L horizon, in RNA or in the DNA.
Ecological functions could be reliably assigned to 64–94% of the abundant members of the fungal community (Supplementary Table 3). Among these, ectomycorrhizal fungi dominated all communities, representing 83% of the sequences in LD, 95% in HD, 66% in LR and 69% in HR (Supplementary Figure 2C). During a period when decomposition processes prevail, a substantial reduction in the activity of ectomycorrhizal fungi compared with saprotrophs is expected (Yarwood et al., 2009; Lindahl et al., 2010). Saprotrophic and parasitic species were indeed significantly more represented in the RNA communities, the ratio of mycorrhizal/saprotrophic+parasitic fungi being 6.9 and 2.9 in the LD and LR and 21.2 and 2.3 in the HD and HR, respectively. Notably, we found many fungal sequences belonging to lichen-forming fungi and detected these preferentially in the RNA community (9.3% in LR).
Ecology of soil bacteria and fungi is largely different
In agreement with previous studies on the vertical stratification of soil decomposition processes (Wittmann et al., 2004; Šnajdr et al., 2008), we show that the L and H horizons differ significantly in both the total and relative amounts of bacterial and fungal biomasses (Figure 1). According to the phospholipid fatty acid/biomass C conversion factors (Anderson and Parkin 2007), the L horizon contained 2.9 times more fungal than bacterial biomass (10.7 and 3.7 mg biomass C per gram, respectively). In the H horizon, fungal and bacterial biomasses were equal (1.50 and 1.58 mg g−1). The diversity of bacterial populations was considerably higher than that of fungal populations. When 1000 randomly selected sequences were analysed, 302–366 OTUs were found in bacteria, compared with 141–236 in fungi.
While the diversity estimates for DNA- and RNA-derived communities of bacteria were similar, the Chao1 estimators for fungal communities surprisingly showed that a more diverse community is revealed when RNA is analysed (Supplementary Table 1). The DNA- and RNA-derived communities of bacteria largely overlapped, and among the abundant OTUs none was found exclusively either in DNA or RNA. By contrast, 18% of fungal OTUs were found only in the RNA community, and 2% were found exclusively in the DNA community. Among cbhI sequences, indicating the presence of cellulolytic members of the fungal community, 27% were found only in DNA and 15% only in RNA (Figure 4). These data show that the DNA sequencing approaches miss a significant and functionally relevant part of microbial communities and our current knowledge largely based on this approach is incomplete. The high RNA/DNA ratios for some microbial taxa show that species with low abundance can be highly active.
Abundances of the major bacterial and fungal OTUs in different soils have been reported to be 2–3% and 7–17%, respectively (Fierer et al., 2007; Buée et al., 2009). In this study, the first to report on bacteria and fungi from the same soil, bacterial communities also showed higher evenness than fungal communities. The most abundant bacterial OTU accounted for 5–7% of all sequences, while the dominant fungal OTU in our ectomycorrhiza-dominated ecosystem represented up to >30% of all sequences. Between 30 and 60 of the most abundant bacterial and 6 and 22 of the most abundant fungal OTUs made up 50% of their respective communities (Supplementary Table 1).
Most bacterial OTUs with abundance >0.3% were recovered from all study sites. In contrast, majority of fungal OTUs and cbhI clusters were found only at one to three sites (Figure 5). Also, the abundance of bacteria across study sites varied considerably less than that of fungi; the mean coefficient of variation was 0.56 for bacteria and 1.30 for fungi. This applied even in the case of the most abundant fungal taxa, for example, the second-most abundant fungal OTU was absent at one study site. The uneven spatial distribution of fungi in the ecosystem is likely a consequence of a combination of factors including the size of organisms, mobility and the association of many taxa with large nutrient patches or plant roots. This is supported by the fact that fungi forming large mycelial systems (for example, Russulales and Agaricales) showed higher variation in abundance than species with limited mycelia (for example, Mortierellales and Archaeosporales).

Number of sites in the Picea abies forest soil where DNA of dominant bacterial and fungal OTUs and cbhI clusters were detected. Only OTUs/clusters with abundance >0.3% in the DNA were considered; n=50 for bacteria, n=54 for fungi and n=56 for cbhI. To correct for the same sampling depth, 350 sequences of each target were randomly selected from each sample.
The affinity of microbial taxa and cbhI clusters for either the L or H horizon and their differential abundance in DNA versus RNA points to their different niches and ecological roles (Supplementary Tables 2–4). Over 60% of dominant fungal OTUs and as much as 74% of cbhI clusters showed 10-fold enrichment in either the L or H horizons. In contrast, vertical stratification was less distinct in bacterial OTUs (Figure 4). This observation further stresses the importance of fungi in shaping the spatial structure of the forest floor.
Cellobiohydrolase genes exhibit high diversity in the soil metagenome and metatranscriptome
Total RNA extracted from soils contains <10% mRNA (Urich et al., 2008); even after mRNA enrichment it still contains both rRNA and transcripts of abundant genes such as those encoding ribosomal proteins (Bailly et al., 2007). This leads to low recovery of targeted sequences of functional genes by shotgun sequencing approaches. For example, only nine gene clusters of denitrification genes were obtained from 77 000 metagenome-derived clones (Demaneche et al., 2009). Amplification of target sequences is thus the only way to assess the diversity of functional genes.
Here, we show for the first time that a single eukaryotic functional gene can be analysed at a depth that allows diversity estimation; a reliable Chao1 diversity prediction of 46±9 cbhI clusters per sample was obtained for the HR sample. When samples from all sites were analysed together, a total of 456 clusters were predicted for LD, 344 for HD, 201 for LR and 99 for HR, with sufficient sampling effort for LR and HR. Approximately 40% and 25% of sequences present in the DNA were being transcribed in the L and H horizons, respectively. Because most of the analysed fungi harbour more than one cbhI gene (Baldrian and Valášková, 2008; Edwards et al., 2008), the diversity of cellulolytic fungi in forest topsoil can be estimated only roughly at 50–300. This means that a considerable proportion of the fungal community transcribes or at least harbours the cbhI gene. Almost all cbhI clusters showed distinct association with either the L or H horizons, indicating only a minor overlap of cellulolytic fungal communities between horizons. Principal component analysis followed by ANOVA showed significant differences in cbhI pool composition among LD, HD, LR and HR (P<0.0019 for differences among L and H and P<0.0054 for DNA/RNA).
Current attempts to assign fungal producers to the sequences of functional genes derived from soil metagenomes suffer from a lack of sequence information in public databases. Because of this, the closest hits for most cbhI clusters in public databases were rather distant, with only three clusters showing >96% similarity. By sequencing cbhI genes from fungi occurring in the soil, we were able to identify the taxonomic affiliation of producers for 13 additional clusters (Supplementary Table 4). Some of the most abundant cbhI sequences were transcribed by fungi with low abundances in the ecosystem (for example, Mycena sp. and Xylariales), showing the importance of low-abundance species for cellulose hydrolysis. Phylogeny trees constructed using cDNA and peptide sequences (Supplementary Figure 3) allowed coarse taxonomic placement of producers for 24–90% of dominant cbhI clusters into either Ascomycota or Basidiomycota (Figure 3). In the H horizon, where 75–90% of sequences were assigned, genes of both taxa were equally present, but the transcripts were mainly of ascomycetous origin (>95% of assigned sequences).
To match the sequences of genes and transcripts, introns were removed from DNA sequences (Supplementary Figure 3). There were 28 DNA clusters containing one intron and one containing four introns. The internal peptide of cbhI obtained after intron removal varied in length and contained both conserved and highly variable regions (Figure 6). The consensus sequence derived from this study differed from the one based on published cbhI sequences from isolated fungal strains at 4 amino-acid positions out of 101 (Edwards et al., 2008). Thus, the depth of environmental amplicon sequencing may contribute to the construction and evaluation of better primers and qPCR probes for targeted functional genes.

Diversity of amino-acid composition and frequency of alternative amino acids over the length of a cbhI internal peptide obtained by translation of the 62 most abundant cbhI sequences detected in Picea abies forest soil. Abundance of alternative amino acids at each position is colour coded; the identity of the most abundant amino acid at each position is indicated, and consensus amino acids (>75%) are highlighted.
Conclusions
Much of what is currently known about the ecology of soil microbial communities has been inferred from studies targeting DNA. Despite similar diversity of microbial communities based on DNA and RNA analysis, the fact that several major fungal OTUs were found exclusively in the RNA pool and that several active bacterial OTUs exhibited low abundance in the DNA pool demonstrates the limitations of DNA-based surveys, which likely miss considerable portions of active microbial populations. In the soil ecosystem, bacterial and fungal communities differ in their spatial distributions with fungal taxa more distinctly confined to either the litter or the organic horizon of soil and more heterogeneously distributed in the ecosystem. The diversity and distribution of functional genes responsible for important biogeochemical processes and consequently of their producers can be efficiently targeted by amplicon sequencing. Low abundance of several fungal taxa highly expressing the cbhI gene suggests that these species are highly important for decomposition.
References
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Amann RI, Ludwig W, Schleifer KH . (1995). Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59: 143–169.
Anderson IC, Parkin PI . (2007). Detection of active soil fungi by RT-PCR amplification of precursor rRNA molecules. J Microbiol Methods 68: 248–253.
Bååth E, Anderson TH . (2003). Comparison of soil fungal/bacterial ratios in a pH gradient using physiological and PLFA-based techniques. Soil Biol Biochem 35: 955–963.
Bailey VL, Smith JL, Bolton H . (2002). Fungal-to-bacterial ratios in soils investigated for enhanced C sequestration. Soil Biol Biochem 34: 997–1007.
Bailly J, Fraissinet-Tachet L, Verner M-C, Debaud J-C, Lemaire M, Wesolowski-Louvel M et al. (2007). Soil eukaryotic functional diversity, a metatranscriptomic approach. ISME J 1: 632–642.
Baldrian P, Valášková V . (2008). Degradation of cellulose by basidiomycetous fungi. FEMS Microbiol Rev 32: 501–521.
Bowker MA, Maestre FT, Escolar C . (2010). Biological crusts as a model system for examining the biodiversity-ecosystem function relationship in soils. Soil Biol Biochem 42: 405–417.
Buée M, Reich M, Murat C, Morin E, Nilsson RH, Uroz S et al. (2009). 454 Pyrosequencing analyses of forest soils reveal an unexpectedly high fungal diversity. New Phytol 184: 449–456.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Demaneche S, Philippot L, David MM, Navarro E, Vogel TM, Simonet P . (2009). Characterization of denitrification gene clusters of soil bacteria via a metagenomic approach. Appl Environ Microbiol 75: 534–537.
Dowd S, Callaway T, Wolcott R, Sun Y, McKeehan T, Hagevoort R et al. (2008). Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8: 125.
Edgar RC . (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26: 2460–2461.
Edwards IP, Upchurch RA, Zak DR . (2008). Isolation of fungal cellobiohydrolase I genes from sporocarps and forest soils by PCR. Appl Environ Microbiol 74: 3481–3489.
Edwards IP, Zak DR . (2010). Phylogenetic similarity and structure of Agaricomycotina communities across a forested landscape. Mol Ecol 19: 1469–1482.
Fierer N, Breitbart M, Nulton J, Salamon P, Lozupone C, Jones R et al. (2007). Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol 73: 7059–7066.
Fulthorpe RR, Roesch LFW, Riva A, Triplett EW . (2008). Distantly sampled soils carry few species in common. ISME J 2: 901–910.
Hawksworth DL . (2001). The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol Res 105: 1422–1432.
Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, Fierer N . (2009). A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3: 442–453.
Kellner H, Vandenbol M . (2010). Fungi unearthed: transcripts encoding lignocellulolytic and chitinolytic enzymes in forest soil. PLoS One 5: e10971.
Kemp PF, Aller JY . (2004). Estimating prokaryotic diversity: when are 16S rDNA libraries large enough? Limnol Oceanogr Methods 2: 114–125.
Korkama T, Pakkanen A, Pennanen T . (2006). Ectomycorrhizal community structure varies among Norway spruce (Picea abies) clones. New Phytol 171: 815–824.
Lauber CL, Hamady M, Knight R, Fierer N . (2009). Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the Continental Scale. Appl Environ Microbiol 75: 5111–5120.
Li W, Godzik A . (2006). Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22: 1658–1659.
Lindahl BD, Ihrmark K, Boberg J, Trumbore SE, Högberg P, Stenlid J et al. (2007). Spatial separation of litter decomposition and mycorrhizal nitrogen uptake in a boreal forest. New Phytol 173: 611–620.
Lindahl BD, de Boer W, Finlay RD . (2010). Disruption of root carbon transport into forest humus stimulates fungal opportunists at the expense of mycorrhizal fungi. ISME J 4: 872–881.
Luis P, Kellner H, Martin F, Buscot F . (2005). A molecular method to evaluate basidiomycete laccase gene expression in forest soils. Geoderma 128: 18–27.
McGuire KL, Treseder KK . (2010). Microbial communities and their relevance for ecosystem models: decomposition as a case study. Soil Biol Biochem 42: 529–535.
Myneni RB, Dong J, Tucker CJ, Kaufmann RK, Kauppi PE, Liski J et al. (2001). A large carbon sink in the woody biomass of Northern forests. Proc Natl Acad Sci USA 98: 14784–14789.
O’Brien HE, Parrent JL, Jackson JA, Moncalvo JM, Vilgalys R . (2005). Fungal community analysis by large-scale sequencing of environmental samples. Appl Environ Microbiol 71: 5544–5550.
Ohm RA, de Jong JF, Lugones LG, Aerts A, Kothe E, Stajich JE et al. (2010). Genome sequence of the model mushroom Schizophyllum commune. Nat Biotechnol 28: 957–963.
Parameswaran P, Jalili R, Tao L, Shokralla S, Gharizadeh B, Ronaghi M et al. (2007). A pyrosequencing-tailored nucleotide barcode design unveils opportunities for large-scale sample multiplexing. Nucleic Acids Res 35: e130.
Reed HE, Martiny JBH . (2007). Testing the functional significance of microbial composition in natural communities. FEMS Microbiol Ecol 62: 161–170.
Reeder J, Knight R . (2010). Rapid denoising of pyrosequencing amplicon data: exploiting the rank-abundance distribution. Nat Methods 7: 668–669.
Roesch LFW, Fulthorpe RR, Riva A, Casella G, Hadwin AKM, Kent AD et al. (2007). Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290.
Rosling A, Landeweert R, Lindahl BD, Larsson KH, Kuyper TW, Taylor AFS et al. (2003). Vertical distribution of ectomycorrhizal fungal taxa in a podzol soil profile. New Phytol 159: 775–783.
Rousk J, Baath E, Brookes PC, Lauber CL, Lozupone C, Caporaso JG et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J 4: 1340–1351.
Strickland MS, Lauber C, Fierer N, Bradford MA . (2009). Testing the functional significance of microbial community composition. Ecology 90: 441–451.
Šnajdr J, Dobiášová P, Větrovský T, Valášková V, Alawi A, Boddy L et al. (2011). Saprotrophic basidiomycete mycelia and their interspecific interactions affect spatial distribution of extracellular enzymes in soil. FEMS Microbiol Ecol, DOI:10.1111/j.1574-6941.2011.01123.x (in press).
Šnajdr J, Valášková V, Merhautová V, Herinková J, Cajthaml T, Baldrian P . (2008). Spatial variability of enzyme activities and microbial biomass in the upper layers of Quercus petraea forest soil. Soil Biol Biochem 40: 2068–2075.
Štursová M, Baldrian P . (2011). Effects of soil properties and management on the activity of soil organic matter transforming enzymes and the quantification of soil-bound and free activity. Plant Soil 338: 99–110.
Tedersoo L, Nilsson RH, Abarenkov K, Jairus T, Sadam A, Saar I et al. (2010). 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol 188: 291–301.
Urich T, Lanzen A, Qi J, Huson DH, Schleper C, Schuster SC . (2008). Simultaneous assessment of soil microbial community structure and function through analysis of the meta-transcriptome. PLoS One 3: e2527.
Wallander H, Johansson U, Sterkenburg E, Durling MB, Lindahl BD . (2010). Production of ectomycorrhizal mycelium peaks during canopy closure in Norway spruce forests. New Phytol 187: 1124–1134.
Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M et al. (2009). Three genomes from the phylum acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75: 2046–2056.
White TJ, Bruns T, Lee S, Taylor J . (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innins MA, Gelfand DH, Sninsky JJ, White TJ (eds). PCR Protocols. Academic Press: San Diego, pp 315–322.
Wilmotte A, Van der Auwera G, De Wachter R . (1993). Structure of the 16 S ribosomal RNA of the thermophilic cyanobacterium chlorogloeopsis HTF (‘mastigocladus laminosus HTF’) strain PCC7518, and phylogenetic analysis. FEBS Lett 317: 96–100.
Wittmann C, Kahkonen MA, Ilvesniemi H, Kurola J, Salkinoja-Salonen MS . (2004). Areal activities and stratification of hydrolytic enzymes involved in the biochemical cycles of carbon, nitrogen, sulphur and phosphorus in podsolized boreal forest soils. Soil Biol Biochem 36: 425–433.
Yarwood SA, Myrold DD, Hogberg MN . (2009). Termination of belowground C allocation by trees alters soil fungal and bacterial communities in a boreal forest. FEMS Microbiol Ecol 70: 151–162.
Acknowledgements
This work was supported by the Czech Science Foundation (526/08/0751), by the Ministry of Education, Youth and Sports of the Czech Republic (LC06066) and by the Institutional Research Concept of the Institute of Microbiology of the ASCR in Prague (AV0Z50200510).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Rights and permissions
About this article
Cite this article
Baldrian, P., Kolařík, M., Štursová, M. et al. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J 6, 248–258 (2012). https://doi.org/10.1038/ismej.2011.95
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.95
Keywords
This article is cited by
-
Water Level Fluctuations Modulate the Microbiomes Involved in Biogeochemical Cycling in Floodplains
Microbial Ecology (2024)
-
Nutrient utilization strategies and age-related changes in Larix principis-rupprechtii forests
Plant and Soil (2024)
-
Stable functional structure despite high taxonomic variability across fungal communities in soils of old-growth montane forests
Microbiome (2023)
-
Compositional Shifts and Assembly in Rhizosphere-Associated Fungal Microbiota Throughout the Life Cycle of Japonica Rice Under Increased Nitrogen Fertilization
Rice (2023)
-
Different bacterial and fungal community patterns in restored habitats in coal-mining subsidence areas
Environmental Science and Pollution Research (2023)
