
Abstract
Reference phylogenies are crucial for providing a taxonomic framework for interpretation of marker gene and metagenomic surveys, which continue to reveal novel species at a remarkable rate. Greengenes is a dedicated full-length 16S rRNA gene database that provides users with a curated taxonomy based on de novo tree inference. We developed a ‘taxonomy to tree’ approach for transferring group names from an existing taxonomy to a tree topology, and used it to apply the Greengenes, National Center for Biotechnology Information (NCBI) and cyanoDB (Cyanobacteria only) taxonomies to a de novo tree comprising 408 315 sequences. We also incorporated explicit rank information provided by the NCBI taxonomy to group names (by prefixing rank designations) for better user orientation and classification consistency. The resulting merged taxonomy improved the classification of 75% of the sequences by one or more ranks relative to the original NCBI taxonomy with the most pronounced improvements occurring in under-classified environmental sequences. We also assessed candidate phyla (divisions) currently defined by NCBI and present recommendations for consolidation of 34 redundantly named groups. All intermediate results from the pipeline, which includes tree inference, jackknifing and transfer of a donor taxonomy to a recipient tree (tax2tree) are available for download. The improved Greengenes taxonomy should provide important infrastructure for a wide range of megasequencing projects studying ecosystems on scales ranging from our own bodies (the Human Microbiome Project) to the entire planet (the Earth Microbiome Project). The implementation of the software can be obtained from http://sourceforge.net/projects/tax2tree/.
Similar content being viewed by others

Introduction
A robust universal reference taxonomy is a necessary aid to interpretation of high-throughput sequence data from microbial communities (Tringe and Hugenholtz, 2008). Taxonomy based on the 16S rRNA gene (16S) is the most comprehensive and widely used in microbiology today (Pruesse et al., 2007; Peplies et al., 2008), but has yet to reach its full potential because numerous microbes belong to taxa that have not yet been characterized and because numerous sequences that could be reliably classified remain unannotated. For example, two thirds of 16S sequences in GenBank are only classified to domain (kingdom), that is, Archaea or Bacteria, by the National Center for Biotechnology Information (NCBI) taxonomy: this taxonomy is likely the most widely consulted 16S-based taxonomy, despite its disclaimer that it is not an authoritative source, in part because classifications are maintained up to date through user submissions. Most of the un(der)classified sequences are from culture-independent environmental surveys; these sequences can swamp BLAST searches, leaving users baffled about the phylogenetic affiliation of their submitted sequences. This shortcoming has been addressed by several dedicated 16S databases, including the Ribosomal Database Project (Cole et al., 2009), Greengenes (DeSantis et al., 2006), SILVA (Pruesse et al., 2007) and EzTaxon (Chun et al., 2007), that classify a higher proportion of environmental sequences. However, improvements are still needed because many sequences remain unclassified and numerous classification conflicts exist between the different 16S databases (DeSantis et al., 2006). Moreover, the emergence of large-scale next-generation sequencing projects such as the Human Microbiome Project (Turnbaugh et al., 2007; Peterson et al., 2009) and TerraGenome (Vogel et al., 2009), and the availability of affordable sequencing to a wide range of users who have traditionally lacked access, mean that the need to integrate new sequences into a consistent universal taxonomic framework has never been greater.
The Greengenes taxonomy is currently based on a de novo phylogenetic tree of 408 135 quality-filtered sequences calculated using FastTree (Price et al., 2010). De novo tree construction is among the most objective means for inferring sequence relationships, but requires either generation of new taxonomic classifications or transfer of existing taxonomic classifications between iterations of trees as the 16S database expands. Previously, we developed a tool that automatically assigns names to monophyletic groups in large phylogenetic trees (Dalevi et al., 2007), which is useful for naming novel (unclassified) clusters of environmental sequences. Here we describe a method to transfer group names from any existing taxonomy to any tree topology that has overlapping terminal node (tip) names. We used this ‘taxonomy to tree’ approach to annotate the 408 135 sequence tree with the NCBI taxonomy as downloaded in June 2010 (Sayers et al., 2011), supplemented with the Greengenes taxonomy from the previous iteration (Dalevi et al., 2007) and CyanoDB (http://www.cyanodb.cz). Explicit rank information, prefixed to group names, was incorporated into the Greengenes taxonomy to help users orient themselves and to improve the consistency of the classification. We assessed the consistency of the resulting classification with the NCBI taxonomy including currently defined candidate phyla (divisions), and present recommendations for consolidation of 34 redundantly named groups and exclusion of one on the basis that its sole representative is chimeric.
Materials and methods
16S data compilation and de novo tree inference
We obtained 16S sequences from the Greengenes database, which extracts these sequences from public databases using quality filters as described previously (DeSantis et al., 2006). We only used sequences that had <1% non-ACGT characters. The sequences were checked for chimeras using UCHIME (http://www.drive5.com/uchime/) and ChimeraSlayer (Haas et al., 2011). We only removed sequences from named isolates if they were classified as chimeric by both tools; we removed other sequences if they were classified as chimeric by either tool or if they were unique to one study, meaning that no similar sequence (within 3% in a preliminary tree) was reported by another study. Quality filtered 16S sequences were aligned based on both primary sequence and secondary structure to archaeal and bacterial covariance models (ssu-align-0.1) using Infernal (Nawrocki et al., 2009) with the sub option to avoid alignment errors near the ends. The models were built from structure-annotated training alignments derived from the Comparative RNA Website (Cannone et al., 2002) as described in detail previously (Nawrocki et al., 2009). The resulting alignments were adjusted to fit the fixed 7682 character Greengenes alignment through identification of corresponding positions between the model training alignments and the Greengenes alignment. Hypervariable regions were filtered using a modified version of the Lane mask (Lane, 1991). A tree of the remaining 408 135 filtered sequences, (tree_16S_all_gg_2011_1) was built using FastTree v2.1.1, a fast and accurate approximately maximum-likelihood method using the CAT approximation and branch lengths were rescaled using a gamma model (Price et al., 2010). Statistical support for taxon groupings in this tree was conservatively approximated using taxon jackknifing, in which a fraction (0.1%) of the sequences (rather than alignment positions) is excluded at random and the tree reconstructed. We use these support values to help guide selection of monophyletic interior nodes for group naming during manual curation.
For evaluation of NCBI-defined candidate phyla, we added 765 mostly partial length sequences, that failed the Greengenes filtering procedure but were required for the evaluation, to the alignment using PyNAST (Caporaso et al., 2010; based on the 29 November, 2010 Greengenes OTU templates) and generated a second FastTree (tree_16S_candiv_gg_2011_1) using the parameters described above.
Transferring taxonomies to trees (tax2tree)
Having constructed de novo trees, the next key step was to link the internal tree nodes to known, named taxa. We used the NCBI taxonomy (Sayers et al., 2011) as the primary taxonomic source to annotate the trees, supplemented by the previous iteration of the Greengenes taxonomy (Dalevi et al., 2007) and cyanoDB (http://www.cyanodb.cz). This taxonomic annotation used a new algorithm called tax2tree. Briefly, tax2tree consists of the following steps:
-
1)
Input consists of a flat file containing the donor taxonomy and an unannotated (no group names) newick format recipient phylogenetic tree with common sequence (tip) identifiers. The tax2tree donor taxonomy is in a very simple format (Supplementary Figure S1) comprising a unique ID followed by a taxonomy string with rank prefixes and was derived from the NCBI taxonomy (ftp://ftp.ncbi.nih.gov/pub/taxonomy/) using a custom Python script. The tax2tree algorithm first filters out non-informative taxonomic assignments from the donor taxonomy strings including the words ‘environmental’, ‘unclassified’, as described previously (Dalevi et al., 2007). After filtering, the remaining assignments at each taxonomic level from domain to species are added to each tip with empty placeholders at levels that are missing taxon names. The result of this phase is a tree in which some of the tips have taxonomic information at some or all ranks, imported directly from a donor taxonomy. In addition, each node in the phylogenetic tree is augmented with a tip start and stop index corresponding to a list that contains tip taxonomy information. This structure allows for rapid lookups of the taxon names present at all of the tips that descend from any internal node.
-
2)
Precision and recall values necessary for the F-measure calculation (see step 5) are calculated and stored on the internal nodes. This caching markedly improves performance on large trees.
-
3)
Each taxonomic rank at each internal node is determined if it is safe to hold a name. A taxonomic rank is considered safe if (a) there exists a name at that taxonomic rank on the tips that descend that is represented on ⩾50% of those tips and (b) the parent taxonomic rank (for example, phylum to class) is also safe. These names are decorated onto the tree at this point resulting in a phylogenetic tree containing many duplicate names on internal nodes.
-
4)
The F-measure (F=2 × ((precision × recall)/(precision+recall))) (van Rijsbergen, 1979) is then calculated for each internal node for each name at each taxonomic rank in order to determine the optimal internal node for a name. The F-measure is defined as the harmonic mean of precision and recall, and balances false positives and false negatives (precision is the fraction of informative tips with a given name under a given node out of the total count of informative tips under the node; recall is the fraction of informative tips descending from a given node out of all the informative tips of the entire tree containing the same name). Node references and F-measure scores are then cached in a 2-dimensional Python dictionary external to the tree keyed by both the rank level and by the taxon name. After all nodes are scored, the 2-dimensional hash table is iterated over and for each unique name, the internal node with the highest F-measure score for each name is retained. Each name will only be saved on an internal node once; all other internal nodes with that name will be stripped of the name. If a tie is encountered, the internal node with the fewest tips is kept. The result of this phase is that the phylogenetic tree contains many names on the internal nodes with each name occurring at most a single time. Gaps in the taxa names decorated onto the tree are likely as the result of polyphyletic groups.
-
5)
Backfilling is used to fill taxonomic gaps in the unique taxon names left on the phylogenetic tree from the F-measure process. For this procedure, the input taxonomy is transformed into a tree and a Python dictionary is constructed that is keyed by the taxon name and valued by its corresponding node. A gap is defined as missing taxonomy rank name information in the phylogenetic tree between a named interior node and its nearest named ancestor (for example, having phylum and order names but without a name for the intervening class rank). For each internal node and nearest named ancestor pair in the phylogenetic tree in which a gap occurs, the taxon name of the node farthest from the root is identified in the input taxonomy. The input taxonomy is traversed until the nearest ancestor's taxa name is found. The names of the nodes traversed in the input taxonomy tree are then appended to the node farthest from the root of the phylogenetic tree. Following the backfilling procedure, it is possible for the phylogenetic tree to have duplicate taxa names.
-
6)
A back-propagation procedure identifies redundant taxon names in the phylogenetic tree that can be collapsed into a single clade. Here, we test whether any internal node has nearest named descendants at a given rank (for example, phylum) that all share the same name. If so, the name can be removed from the descendants and propagated to the internal phylogenetic node being interrogated.
Secondary taxonomies were then applied manually to the annotated recipient tree, tree_16S_candiv_gg_2011_1, in ARB (Ludwig et al., 2004) using the group tool. For the Greengenes taxonomy, this was achieved by displaying the Greengenes taxonomy field at the tips of the tree and assigning group names missed in the automated taxonomy transfer (mostly higher level ranks associated with candidate phyla). For the cyanoDB taxonomy, manual assignments were based on type species (http://www.cyanodb.cz/valid_genera; 405 instances in tree_16S_candiv_gg_2011_1; Supplementary Table S1). The manually supplemented taxonomic assignments were then exported from the curated tree as a flat file (Supplementary Figure S1) using functionality in the tax2tree pipeline and reapplied to tree_16S_all_gg_2011_1 ensuring manual updates were efficiently propagated to both trees. The tax2tree software is implemented in the Python programming language using the PyCogent toolkit (Knight et al., 2007), and is available under the open-source General Public license at http://sourceforge.net/projects/tax2tree/.
Taxonomy comparisons
The NCBI and Greengenes taxonomies were compared for each of the 408 135 sequences in tree_16S_all_gg_2011_1 making use of the explicit rank designations. The lowest classified rank for each sequence was determined and compared (Figure 2a) to estimate overall improvements in classification. Note that only contiguous classifications were used in this estimate, that is, all ranks leading to the lowest named rank also had to have names. Taxonomic similarities and differences for each sequence at each rank were also assessed by dividing sequences into five categories, (i) the two taxonomies had equal values (same name) at a given taxonomic rank, (ii) the two taxonomies had unequal values at a given rank, (iii) and (iv) one of the taxonomies lacked a value at a given rank and (v) both taxonomies lacked a value at a given rank. This provided an indication of the type of changes that had occurred between the NCBI and Greengenes taxonomies (Figure 2b).
Results and discussion
The rapid accumulation of sequence data from across the tree of life is a boon for molecular taxonomy, but also presents a major barrier to sequence-based taxonomy curators as it is essentially impossible to manually curate trees comprising hundreds of thousands of sequences from scratch. We overcame this difficulty by developing an automated procedure based on F-measures (van Rijsbergen, 1979) for transferring any (donor) taxonomy (in a standard flat text format, Supplementary Figure S1) to any unannotated (recipient) tree (in Newick format) given common sequence identifiers. The F-measure is most often used to measure the classification performance (precision and recall) of information retrieval processes such as database searches (van Rijsbergen, 1979). This approach also has the potential to provide an assessment of the quality of fit between a taxonomy and a tree, which could be used to screen multiple taxonomies and/or trees before manual curation efforts.
Construction of the rank-explicit Greengenes taxonomy
Using quality-filtered sequences from Greengenes (DeSantis et al., 2006) aligned with the secondary structure-aware infernal package (Nawrocki et al., 2009), we constructed a phylogenetic tree using FastTree2 (Price et al., 2010) containing 408 135 sequences (tree_16S_all_gg_2011_1). We inferred confidence estimates using taxon jackknife resampling (in which sequences, rather than positions in the alignment, are resampled) as it provides a conservative guide to group monophyly, which we found greatly assists manual group name curation between tree iterations (see below). We then applied NCBI classifications to this topology using the tax2tree algorithm (Figure 1 and methods) also taking advantage of the explicit rank designations provided by NCBI to include rank prefixes to group names (for example, p(hylum)__, c(lass)__, o(rder)__). Most sequences (69%; 280 488 of 408 135) had uninformative NCBI classifications, that is, no rank information below domain (kingdom; Figure 2a); of these, most were environmental clones designated as ‘unclassified Bacteria’. The remaining 127 647 sequences with informative classifications were then applied to the tree. This ‘unamended’ approach alone resulted in an improved classification, to at least phylum-level, of nearly all of the taxonomically uninformative sequences (280 452 of 280 488) because most belong to known phyla but were simply deposited without classifications.

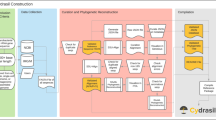
Overview of the tax2tree workflow. (i) The inputs to tax2tree; a taxonomy file that matches known taxonomy strings to identifiers that are associated with tips of (that is, sequences within) a phylogenetic tree. To simplify the diagram, only the family, genus and species are used, although the full algorithm uses all phylogenetic ranks. (ii) The input taxonomy represented as a tree and a taxon name legend for the figure. (iii, iv) Nodes chosen by the F-measure procedure at each rank; (iii) species, (iv) genus and (v) family. In this example, the genus Clostridium is polyphyletic, and the F-measure procedure picked the ‘best’ internal node for the name (uniting tips A–F). However, as unique names at a given rank can only be placed once on the tree, this leaves tips I–L without a genus name placed on an interior node. (vi) The backfilling procedure detects that tips I–L have an incomplete taxonomic path (species to family) and (vi) prepends the missing genus name (obtained from the input taxonomy) to the lower rank because this step of the procedure examines only ancestors but not siblings. (vii) The common name promotion step identifies internal nodes in which all of the nearest named descendants share a common name. In this example, the node that is the lowest common ancestor for tips I–L has immediate descendants that all share the same genus name, Clostridium. This name can be safely promoted to the lowest common ancestor (interior node) uniting tips I–L. (viii) The resulting taxonomy. Note that the sequence identified as B was unclassified in the donor taxonomy but is now classified as f__Lachnospiraceae; g__Clostridium; s__.

A comparison of the NCBI taxonomy to the updated Greengenes taxonomy for sequences in tree_16S_all_gg_2011_1. (a) Lowest taxonomic rank assigned to each sequence; (b) taxonomic differences between NCBI and Greengenes at each rank, showing the percentage of sequences classified to each of five possible categories (see inset legend; GG, Greengenes) highlighting cases where NCBI and Greengenes differ.
We then overlaid additional taxonomic information onto the NCBI-annotated tree by manual group name curation in ARB. This information consisted primarily of candidate phyla and other rank designations for environmental clusters imported from previous iterations of Greengenes (either assigned manually or by GRUNT (Dalevi et al., 2007)), and taxonomic information for the Cyanobacteria obtained from cyanoDB (http://www.cyanodb.cz/). This resulted in more informative classifications for 75% of sequences by at least one rank up to six ranks. These increases in classification depth are graphically shown in Figure 2a by rank.
Changes in sequence classifications between NCBI and Greengenes at each rank are summarized in Figure 2b. Most changes were from uninformative (domain/kingdom name only) in NCBI to informative in Greengenes again reflecting the classification of the large fraction of unclassified environmental sequence in NCBI. The percentage of changes to informative NCBI classifications were relatively low (<7% for all ranks), indicating the degree of congruence between NCBI and Greengenes classifications achieved in part by accommodating polyphyletic groups (see below). Of these type of changes, many occurred at the higher ranks particularly in the candidate phyla where Greengenes is manually curated most intensively (see below). A similar comparison with SILVA or RDP was not possible because of a lack of explicit ranks in these taxonomies. However, by comparing the group name immediately following the domain (kingdom) name (either Bacteria or Archaea) in the SILVA taxonomy, we estimated that only 28% of the 408 135 sequences lacked phylum-level classifications in SILVA, as opposed to 68% in NCBI.
Further, the updated Greengenes taxonomy performed well in a test of reference taxonomies using a naïve Bayesian classifier (Werner et al., 2011). In this paper, we found that retraining the RDP classifier (Wang et al., 2007) using taxa from the new Greengenes taxonomy resulted in increased classification resolution relative to SILVA or RDP for a range of environments (human body habitats, snake and mouse gut and soils).
The value of accommodating polyphyletic groups in a 16S rRNA-based taxonomy
In principle, every taxon should correspond to a single monophyletic group in the 16S rRNA-based taxonomy, but practical considerations make relaxing this constraint very useful. In our approach, the back-filling step in the tax2tree (see methods) allows multiple groups to be given the same rank name. This feature is important for taxonomic groups that are well-established in the literature but polyphyletic in the reference 16S rRNA tree. A prominent example is the class Deltaproteobacteria, which rarely forms a monophyletic group in large 16S rRNA topologies and comprises five groups in the current tree_16S_all_gg_2011_1. This result may indicate that the Deltaproteobacteria do not form an evolutionary coherent grouping and will need to subdivided and reclassified. Alternatively, the Deltaproteobacteria may be a monophyletic group not resolved in 16S rRNA trees due to tree inference artifacts, chimeric sequences and/or to limits in the phylogenetic resolution of trees constructed from the 16S rRNA molecule alone. This issue can best be addressed using ‘whole genome’ tree approaches that have greater phylogenetic resolution than single-gene topologies. Trees based on a concatenation of 31 conserved near ubiquitous single-copy gene families indicate that the small subset of Deltaproteobacteria with genome sequences are monophyletic (Ciccarelli et al., 2006; Wu et al., 2009). A second example, also based on concatenated conserved marker genes, indicate that the first genome sequence representative of the family Halanaerobiales (Halothermothrix orenii) is a member of the Firmicutes phylum (Mavromatis et al., 2009); which is its (contested) classification based on 16S rRNA trees (Ludwig and Klenk, 2001). Indeed, the Halanaerobiales is separate from the Firmicutes in the current Greengenes topology and is only classified as such because of the back-filling procedure.
Similarly, a number of phylum-level associations have been suggested based on concatenated gene topologies including a relationship between the class Deltaproteobacteria and phylum Acidobacteria (Ciccarelli et al., 2006). We predict that at least a subset of the currently defined candidate phyla will coalesce with other phyla once they are adequately represented by genome sequences and whole-genome trees can be constructed. Therefore, current estimates of the number of 16S-based candidate phyla (>50) should only be used as an approximation and may well drop as candidatus genome sequences accrue. However, regardless of absolute number of phyla, there is a strong need for consistent delineation of taxonomic groups between public databases, particularly candidate phyla. Thus, by retaining polyphyletic groups as sets of monophyletic taxa with the same name, we can accommodate the uncertainty in our present knowledge about both the tree and taxonomy and easily propagate whole-genome-based classification improvements in subsequent iterations of de novo 16S rRNA-based trees.
Reconciliation of NCBI and Greengenes-defined candidate phyla
At the time of writing, the NCBI taxonomy lists 71 candidate phyla (divisions) of which 30 are represented only by partial (<1200 nt) sequences. Therefore, in order to address the classification of these groups we amended tree_16S_all_gg_2011_1 with 765 mostly partial length sequences from GenBank and generated a new de novo tree using FastTree; tree_16S_candiv_gg_2011_1. We found some NCBI candidate phyla to be polyphyletic in tree_16S_candiv_gg_2011_1 because of a small number of submitter misclassifications or chimeric artifacts. In these instances, we reconciled NCBI and Greengenes designations using the majority classification for a given NCBI group. On the basis of tree_16S_candiv_gg_2011_1, we resolved the 71 candidate phyla into 45 monophyletic groups and one chimeric artifact (Table 1). Many proposed phyla appear to belong to well-established lineages including the Proteobacteria, Firmicutes, Bacteroidetes, Chloroflexi and Spirochaetes. In cases where two or more NCBI candidate phyla were combined and did not cluster with more established groups, we gave priority to either the oldest and/or the largest group. For example, we reclassified candidate phylum kpj58rc (Kelly and Chistoserdov, 2001) as OP3 (Hugenholtz et al., 1998) because of the priority of OP3 in the literature and larger number of representative OP3 sequences in the public databases. We also compared our classifications to SILVA and RDP and in many instances saw consistencies. For example, Greengenes and SILVA both classify candidate phylum CAB-I as Cyanobacteria and Greengenes and RDP both classify KSA1 in the Bacteroidetes. Conversely, in some instances we saw disagreements, such as candidate phylum GN02 (Ley et al., 2006) being classified as BD1-5, and WS5 (Dojka et al., 1998) as WCHB1-60 by SILVA (Table 1). This points to the need to consolidate classifications and also to give priority to published group names where possible.
Final comments and prospectus
The new NCBI-reconciled Greengenes taxonomy rescues over 200 000 environmental sequences from unclassified oblivion. Moreover, the tax2tree pipeline will assist in reconciling information among the various 16S rRNA resources (Greengenes, SILVA, RDP and EZ-Taxon) with phylogenetic trees built using different methods, and will, we hope, make it easier for users to reconcile taxonomic classifications of large data sets obtained using different taxonomic schemes. This is especially important because which taxonomy is used can have a larger effect on the results than which assignment method is used (Liu et al., 2008). The new Greengenes taxonomy, along with all intermediate data products including the tree, can be downloaded from the Greengenes web site at http://greengenes.lbl.gov/.
This work, by automating the process of improving the tree and allowing import of taxonomic knowledge from elsewhere, provides the first step toward an automated pipeline that will immensely improve our ability to link organisms to environment and to understand the evolutionary change associated with phenotypic changes such as adaptation to a new host, switching to a new habitat or adapting to use a new substrate. By providing the foundation for organizing microbial knowledge, these expanded taxonomies will greatly expand our ability to understand the microbes that pervade all aspects of life on the Earth.
References
Cannone JJ, Subramanian S, Schnare MN, Collett JR, D’Souza LM, Du Y et al. (2002). The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform 3: 2.
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R . (2010). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26: 266–267.
Chun J, Lee JH, Jung Y, Kim M, Kim S, Kim BK et al. (2007). EzTaxon: a web-based tool for the identification of prokaryotes based on 16S ribosomal RNA gene sequences. Int J Syst Evol Microbiol 57: 2259–2261.
Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P . (2006). Toward automatic reconstruction of a highly resolved tree of life. Science 311: 1283–1287.
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37: D141–D145.
Dalevi D, DeSantis TZ, Fredslund J, Andersen GL, Markowitz VM, Hugenholtz P . (2007). Automated group assignment in large phylogenetic trees using GRUNT: GRouping, Ungrouping, Naming Tool. BMC Bioinform 8: 402.
DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72: 5069–5072.
Dojka MA, Hugenholtz P, Haack SK, Pace NR . (1998). Microbial diversity in a hydrocarbon- and chlorinated-solvent-contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64: 3869–3877.
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G et al. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21: 494–504.
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR . (1998). Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180: 366–376.
Kelly KM, Chistoserdov AY . (2001). Phylogenetic analysis of the succession of bacterial communities in the Great South Bay (Long Island). FEMS Microbiol Ecol 35: 85–95.
Knight R, Maxwell P, Birmingham A, Carnes J, Caporaso JG, Easton BC et al. (2007). PyCogent: a toolkit for making sense from sequence. Genome Biol 8: R171.
Lane DJ . (1991). 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds). Nucleic Acid Techniques in Bacterial Systematics. John Wiley and Sons: West Sussex.
Ley RE, Harris JK, Wilcox J, Spear JR, Miller SR, Bebout BM et al. (2006). Unexpected diversity and complexity of the Guerrero Negro hypersaline microbial mat. Appl Environ Microbiol 72: 3685–3695.
Liu Z, DeSantis TZ, Andersen GL, Knight R . (2008). Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 36: e120.
Ludwig W, Klenk H-P . (2001). Overview: a phylogenetic backbone and taxonomic framework for procaryotic systematics. In: Boone DR, Castenholtz RW, Garrity GM (eds). Bergey's Manual of Systematic Bacteriology. Springer: New York.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Kumar Y et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Mavromatis K, Ivanova N, Anderson I, Lykidis A, Hooper SD, Sun H et al. (2009). Genome analysis of the anaerobic thermohalophilic bacterium Halothermothrix orenii. PloS One 4: e4192.
Nawrocki EP, Kolbe DL, Eddy SR . (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25: 1335–1337.
Peplies J, Kottmann R, Ludwig W, Glockner FO . (2008). A standard operating procedure for phylogenetic inference (SOPPI) using (rRNA) marker genes. Syst Appl Microbiol 31: 251–257.
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA et al. (2009). The NIH Human Microbiome Project. Genome Res 19: 2317–2323.
Price MN, Dehal PS, Arkin AP . (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PloS One 5: e9490.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J et al. (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 35: 7188–7196.
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K et al. (2011). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39: D38–D51.
Tringe SG, Hugenholtz P . (2008). A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol 11: 442–446.
Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI . (2007). The human microbiome project. Nature 449: 804–810.
van Rijsbergen CV . (1979). Information Retrieval 2nd edn. Butterworth: Boston.
Vogel TM, Simonet P, Jansson JK, Hirsch PR, Tiedje JM, van Elsas JD et al. (2009). TerraGenome: a consortium for the sequencing of a soil metagenome. Nat Rev Micro 7: 252.
Wang Q, Garrity GM, Tiedje JM, Cole JR . (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267.
Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG et al. (2011). Impact of training sets on classification of high-throughput bacterial 16S rRNA gene surveys. ISME J; e-pub ahead of print 30 June 2011, doi: 10.1038/ismej.2011.82.
Wu D, Hugenholtz P, Mavromatis K, Pukall R, Dalin E, Ivanova NN et al. (2009). A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462: 1056–1060.
Acknowledgements
We thank Adam Arkin and Sean Eddy for supporting this study. The work conducted by ENIGMA was supported by the Office of Science, the Office of Biological and Environmental Research, of the US Department of Energy under Contract no. DE-AC02-05CH11231. This work was supported in part by the Sloan Indoor Environment program, the National Institutes of Health (grants UH2/UH3CA140233 and U01-HG004866), the Bill and Melinda Gates Foundation and the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
McDonald, D., Price, M., Goodrich, J. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618 (2012). https://doi.org/10.1038/ismej.2011.139
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2011.139
Keywords
This article is cited by
-
High-throughput identification and quantification of bacterial cells in the microbiota based on 16S rRNA sequencing with single-base accuracy using BarBIQ
Nature Protocols (2024)
-
Defined microbial communities and their soluble products protect mice from Clostridioides difficile infection
Communications Biology (2024)
-
A genus in the bacterial phylum Aquificota appears to be endemic to Aotearoa-New Zealand
Nature Communications (2024)
-
Mock community taxonomic classification performance of publicly available shotgun metagenomics pipelines
Scientific Data (2024)
-
Metagenetic analysis of the bacterial diversity of Kazakh koumiss and assessment of its anti-Candida albicans activity
World Journal of Microbiology and Biotechnology (2024)
