
Abstract
Microbial biogeographic patterns in the deep sea depend on the ability of microorganisms to disperse. One possible limitation to microbial dispersal may be the Walvis Ridge that separates the Antarctic Lower Circumpolar Deep Water from the North Atlantic Deep Water. We examined bacterial communities in three basins of the eastern South Atlantic Ocean to determine diversity and biogeography of bacterial communities in deep-sea surface sediments. The analysis of 16S ribosomal RNA (rRNA) gene clone libraries in each basin revealed a high diversity, representing 521 phylotypes with 98% identity in 1051 sequences. Phylotypes affiliated with Gammaproteobacteria, Deltaproteobacteria and Acidobacteria were present in all three basins. The distribution of these shared phylotypes seemed to be influenced neither by the Walvis Ridge nor by different deep water masses, suggesting a high dispersal capability, as also indicated by low distance–decay relationships. However, the total bacterial diversity showed significant differences between the basins, based on 16S rRNA gene sequences as well as on terminal restriction fragment length polymorphism fingerprints. Noticeably, both geographic distance and environmental heterogeneity influenced bacterial diversity at intermediate (10–3000 km) and large scales (>3000 km), indicating a complex interplay of local contemporary environmental effects and dispersal limitation.
Similar content being viewed by others


Introduction
Biogeographic patterns in microbial communities are traditionally explained by two factors, the environmental heterogeneity and historical events (Martiny et al., 2006; O’Malley, 2008). On the basis of the cosmopolitan hypothesis, ‘everything is everywhere, but the environment selects’ (Baas-Becking, 1934), environmental conditions have long been considered to have a strong influence on microbial biogeography. The effects of spatial distances (historical events) have been shown to affect microbial diversity in several studies (Papke et al., 2003; Whitaker et al., 2003; Martiny et al., 2006; Ramette and Tiedje, 2007). The relative influences of environmental heterogeneity and historical events on microbial biogeography are still poorly understood. In marine habitats like the deep sea, microorganisms in the surface sediment may be assumed to disperse with oceanic currents. Bioirrigation by the activities of larger benthic organism as well as near-bed currents (Hughes and Gage, 2004; Queric and Soltwedel, 2007) influence the sediment-water interface exchange and consequently lead to the dispersal of particles and therefore of microorganism. Barriers to microbial dispersal could be physical (topography) or physiological conditions (temperature, pH or hydrostatic pressure).
In the eastern South Atlantic Ocean, the Cape Basin is separated from the Angola and Guinea basins by the Walvis Ridge that forms a barrier to the northward and southward flow of water below a depth of about 3000 m (Shannon and Chapman, 1991). Furthermore, the Cape Basin is dominated by Lower Circumpolar Deep Water arriving from Antarctica and the deepest part of the Angola and Guinea Basins are filled with North Atlantic Deep Water originating from the Arctic (Bickert and Wefer, 1996). Noticeably, the Walvis Ridge has been shown to function as a barrier for the dispersal of some crustacean species of Peracarida (Brandt et al., 2005), but it is not known whether this physical barrier also affects microbial dispersal.
To analyze whether different deep water masses associated with the physical barrier of the Walvis Ridge have significant structuring effects on microbial diversity, the bacterial diversity in three deep-sea basin surface sediments was determined by 16S ribosomal RNA (rRNA) gene sequencing and the community fingerprinting method terminal restriction fragment length polymorphism (T-RFLP). The relative contribution of environmental heterogeneity and of historical events on microbial biogeography were assessed for this data set in concert with earlier published data on basaltic lavas in the Pacific Ocean (Santelli et al., 2008), shallow permanently cold sediment of the Arctic Ocean (Ravenschlag et al., 1999) and Antarctic continental shelf sediment (Bowman and McCuaig, 2003).
Materials and methods
16S ribosomal RNA gene clone libraries construction
Sediment sampling was performed on the DIVA II cruise by a multicorer (Barnett et al., 1984) in water depths ranging from 5032 to 5649 m. The sediment cores were sliced on board in layers of 2 cm and the layers were subsampled top-to-bottom by sterile 1- to 2-ml syringes at 4 °C. After storage at −80 °C, DNA was extracted from 0.5 g of the surface sediment sample (0–2 cm) of the Cap, Angola and Guinea I areas (Figure 1, Table 2) after the protocol of the FastDNA SPIN Kit for Soil (Q-BIOgene, Carlsbad, CA, USA). Bacterial 16S rRNA genes were amplified using the primer pair GM3/GM4 (Muyzer et al., 1995). The 100-μl reaction contained 30 ng DNA as template, 0.5 μM of each primer, 10 mM of dNTPs, 1 × buffer (Eppendorf, Hamburg, Germany) and 5 U of the Takara-Taq DNA polymerase (TAKARA, Dalian, China). PCRs were performed in 10 replicates with 20 cycles to minimize PCR bias. Final extension was performed 60 min at 60 °C to increase 3′-A-overhang. The amplicons were pooled and purified with a PCR purification kit (Qiagen, Hilden, Germany). Cloning of the amplicons was performed using TOPO TA Cloning Kit for sequencing (pCR4-TOPO, Invitrogen, Karlsruhe, Germany). Clones with a correct insert size of ∼1500 bp were sequenced using the vector primers M13 F (5′-GGAAACAGCTATGACCATG-3′) and M13 R (5′-GTTGTAAAACGACGGCCAGT-3′).

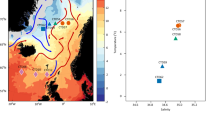
Sampling areas in the South Atlantic Ocean as well as the Walvis Ridge that separates the Cape Basin from the Angola and Guinea basins. For the 16S ribosomal RNA (rRNA) gene approach surface sediment (0–2 cm) of the Cape, Angola and Guinea I areas were used and for the terminal restriction fragment length polymorphism (T-RFLP) analysis 3–5 surface sediments of the Cape, Angola and Guinea I–III areas were analyzed.
Phylogenetic and sequence analyses
The quality of the obtained sequences was manually checked using Sequence Analysis 5.2 (Applied Biosystems, Weiterstadt, Germany). Full-length sequences were assembled with Sequencher (Gene Code, Ann Arbor, MI, USA). No chimeras were detected with Bellerophon (Huber et al., 2004) and CHECK_CHIMERA (Maidak et al., 1996). Sequences were imported into the ARB software package (Ludwig et al., 2004) and aligned using the ARB FastAligner, then refined manually. The ARB software package was used to generate phylogenetic trees of 810 full-length sequences using the maximum likelihood algorithm with a 50% positional conservation filter and with 100 bootstrap replicates. Sequences reported in this study were deposited at EMBL under the accession numbers AM997284–AM997988 for 705 full-length sequences and under AM997989–998333 and AM997283 for 346 partial sequences.
The software distance-based OTU and richness (DOTUR) was applied to ARB distance matrices generated with the Jukes-Cantor correction to estimate operational taxonomic units (OTU), rarefaction curves of observed OTUs, richness estimators and diversity indices (Schloss and Handelsman, 2005). A sequence identity of 98% was used to define OTUs, as this cut-off roughly corresponds to the species level (Rossello-Mora and Amann, 2001; Stackebrandt and Ebers, 2006). The statistical tool ∫-LIBSHUFF was applied to genetic distance matrices to determine whether differences in library composition were because of chance or to biological effects, and significances were assessed by Monte Carlo permutations and further corrected for multiple comparisons (Schloss et al., 2004). The statistical tool SONS (Schloss and Handelsman, 2006) was used on full-length 16S rRNA gene sequences to calculate Chao1 shared richness estimates, the Jclass index for the ratio of shared to total number of OTUs, and θyc for the estimated similarity in community structure between any two communities.
Terminal restriction fragment length polymorphism
Terminal restriction fragment length polymorphism analyses included three to five samples of surface sediments (0–2 cm) from several cores of each area, Cape, Angola and Guinea I–III (Figure 1, Table 2). Genomic DNA was extracted from 0.5 g sediment samples using the FastDNA Spin Kit for Soil (Q-Biogene, Irvine, CA, USA). PCR amplification of the 16S rRNA gene was carried out using the fluorescently labelled primers 27F (FAM, 5′-AGAGTTTGA TCCTGGCTCAG-3′) and 907R (HEX, 5′-CCGTCAATTCCTTTRAGTTT-3′), targeting all bacteria as well as 558F (FAM, 5′-ATTGGGTTTAAAGGGTCCG-3′) (Abell and Bowman, 2005a, 2005b) and 1390R (HEX, 5′-GACGGGCGGTGTGTACAA-3′) (Zheng et al., 1996), targeting the class Flavobacteria. Undigested and digested amplicons were identified by capillary electrophoresis to verify the absence of false-positive fragments in the undigested control and the completeness of the digestion. PCRs were carried out in a total volume of 25 μl, including 12.5 μl PCR Master Mix (Promega GmbH, Mannheim, Germany), 1 μM forward and reverse primer, and 5–24 ng DNA template. PCR reactions were carried out in triplicates and purified on Sephadex columns (Sephadex G-50 Superfine, Amersham Biosciences AB, Uppsala, Sweden). PCR amplicons (70-120 ng) were digested in a total volume of 10 μl at 37 °C for 3 h using 5 U of the restriction enzyme AluI (Fermentas, Burlington, Canada) for bacterial amplicons and 5 U of the enzyme MspI (Fermentas) for Flavobacteria amplicons. The two restriction enzymes were chosen based on high numbers of unique terminal restriction fragments assessed with in silico analyses using enzyme restriction power analysis (http://mica.ibest.uidaho.edu/) as well as on best performance in laboratory experiments (that is, producing maximum numbers of terminal restriction fragments (TRFs)). After heat inactivation (65 °C, 25 min) and purification on Sephadex columns, detection of TRFs was performed on a ABI Prism 3130 XL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) equipped with a 80-cm capillary, a POP-7 polymer and the filter set DS-30. The ROX-labelled MapMarker 1000 (Eurogentec, Seraing, Belgium) served as a size standard between 50 and 1000 bp. The fragment profiles were visualized and automatically analyzed with GeneMapper v. 3.7 Software (Applied Biosystems), using standardized settings with a peak detection cut-off set to 30 fluorescence units. The 5′-end labelled TRFs were used as they produced a higher number of fragments in comparison with 3′-end TRFs (Suzuki et al., 1998; Osborn et al., 2000).
A binning procedure was applied to the GeneMapper output to compensate for slight peak shifts between runs and for TRF size calling imprecision, in order to avoid artificial, technically derived differences between profiles (Hewson and Fuhrman, 2006). The technical variability of peak size calling in different replicates including runs conducted on different days was determined as of±0.25 bp (window size of 0.5 bp). The binning function included two different starting points (50 and 50.25 bp) and the binning strategy yielding higher correlation between all samples was selected for further statistical analyses. The binning window was adjusted to 1 bp for samples amplified with Flavobacteria primers, because a window frame of 0.5 bp did not yield higher resolution. The computation was carried out with the Interactive Binner function (Ramette, 2009 http://www.ecology-research.com). The output consisted of a table of TRFs with corresponding relative fluorescence intensities, which are the individual peak area divided by the total area of peaks in a given profile. Master profiles were generated by building a consensus table of the binned TRF profiles for all samples from one basin, averaging the respective relative fluorescence intensities values of all samples. A TRF was considered present if it appeared in one or more PCR parallels, therefore including all natural and technical variability at this level of analysis.
Statistical analyses
Non-metric multidimensional scaling (nMDS) and analysis of similarity (ANOSIM) were carried out with the program PAST (Paleontological Statistics, ver. 1.47, http://folk.uio.no/ohammer/past). Simple and partial Mantel tests were used to determine the significance and correlation coefficients between genetic-, spatial- and environmental distance matrices, using the R package vegan (http://vegan.r-forge.r-project.org/) (Legendre and Legendre, 1998; Mantel, 1967). Spatial dissimilarities based on geographic distances between sites and environmental dissimilarities (temperature, salinity, pH, Eh, TOC, Chl a and grain size; Table 1) were used to explain genetic dissimilarity. To determine the strength of the relationship between genetic and geographic distance linear models were fitted and slope coefficients were calculated with their 95% confidence intervals.
Results and discussion
Bacterial biomass and richness in sediments of the South Atlantic Ocean
The cell numbers of the suboxic surface sediments (0–2 cm) in three eastern South Atlantic Ocean basins were 3.4–3.7 × 109 cells g–1 sediment (Table 1). The abundances were in the range found in other deep-sea sediments (9.2 × 108 cells g–1 (Deming and Colwell, 1982), 1.5 × 109 cells g–1 (Guezennec and Fiala-Medioni, 1996) and 5 × 108 cells g–1 (Harvey et al., 1984)). The 16S rRNA gene libraries showed a high diversity with up to 20 different phyla in the Cape Basin and 17 phyla in the Angola and Guinea basins (Figure 1). Earlier described deep-sea (Bowman and McCuaig, 2003; Polymenakou et al., 2005, 2009; Xu et al., 2005) and shallow sediments (Ravenschlag et al., 1999) had also found a large diversity, which may be based on a weak and symmetric competition (Grant, 2000). The reciprocal Simpson's indices for all sites were above 50 (Table 2), suggesting evenly distributed diversity profiles as typical dominance profiles show 1/D values below approximately 50 (Zhou et al., 2002). Total richness estimates (Chao1) (Table 2) and rarefaction curves (Supplementary Figure S1) based on a 98% sequence identity showed that Cape, Angola and Guinea basin surface sediments contained an equal bacterial richness at a significance level of 0.05.
Both analyses predicted a lower richness for the South Atlantic sediments in comparison to the Antarctic sediments and a higher richness in comparison to the Arctic sediment. The library-based equality of richness was supported by the T-RFLP analysis, as basin-specific master profiles showed a comparable OTU richness (167, 190 and 182 TRFs for the Cape, Angola and Guinea Basin, respectively) (Figure 4a).
Bacterial diversity of the 16S ribosomal RNA genes
The clone libraries contained 521 phylotypes with 98% identity in 1051 sequences, containing 705 full-length sequences. Applying a 100% identity threshold revealed 230 sequences, which were present at least twice, with a majority of 176 sequences (18 OTUs) present in all deep-sea sediments. The bacterial communities were dominated by Proteobacteria, which accounted for 64, 58 and 63% of all sequences in the Cape, Angola and Guinea Basin, respectively, with the class Gammaproteobacteria representing 45, 37 and 40% of all sequences in the respective basins (Figure 2). The class Gammaproteobacteria comprised 116 phylotypes (98% identity, 427 sequences), of which 39 phylotypes (138 sequences) were related to known cultivated species. These belonged mainly to families of psychrophilic microorganisms including Enterobacteriaceae, Alteromonadaceae, Oceanospirillaceae and Legionellaceae (Figure 3a). Among these phylotypes 11 OTUs (12 sequences) clustered with the NOR5/OM60 clade that includes ‘Congregibacter litoralis’ strain KT71, the first marine aerobic anoxygenic phototrophic Gammaproteobacteria in culture (Fuchs et al., 2007; Yan et al., 2009). Three phylotypes (5 sequences) were related to free living (Thiothrix) and endosymbiotic sulfur oxi'dizers and methylotrophic bacteria. A large portion of 77 phylotypes (289 sequences) clustered distinctly from cultured species to JTB255/BD3-6 (38 phylotypes, 192 sequences), BD7-8/MERTZ (10 OTUs, 36 sequences), JTB23/Sva0091 (18 OTUs, 34 sequences) (Figures 3a and b) and to Cret-1F, BD1-1, PWP and South Ionian groups (11 OUT, 27 sequences). These groups included only 16S rRNA gene sequences that originated from other deep-sea or permanent cold marine habitats (Kato et al., 1999; Li et al., 1999; Ravenschlag et al., 1999; Urakawa et al., 1999; Bowman and McCuaig, 2003; Polymenakou et al., 2005; Xu et al., 2005; Zhao and Zeng, 2005).

Bacterial diversity in the Cape Basin (342 sequences), Angola Basin (354 sequences) and Guinea Basin (355 sequences). All detected classes in the domain Bacteria are shown.

Phylogenetic tree based on 16S ribosomal RNA (rRNA) gene sequences of the class Gammaproteobacteria showing position of (a) marine heterotrophic aerobic and facultative anaerobic microorganisms and photoheterotrophic aerobic (NOR5/OM60 clade) bacteria and (b) potential auto- or mixotrophic sulfur oxidizers and bacteria that inhabit various geographic regions (JTB255/BD3-6). The tree was calculated using the maximum-likelihood algorithm with a 50% positional conservation filter and with 100 bootstrap replicates. The bar represents 10% estimated sequence divergence. Full-length sequences (Ca, An and Gu), partial sequences (cap, ang and gui), the number of OTUs in a cluster and the corresponding number of sequences (squared bracket) are shown.
The Alpha-, Beta- and Deltaproteobacteria accounted together for 18 to 23% of all sequences in the libraries. Deltaproteobacteria (11 to 14%) outnumbered Alphaproteobacteria (6 to 8%) and Betaproteobacteria (1 to 3%) (Figure 2). Other groups with a sequence abundance of over 5%, which occurred in all three basins, were the phyla Chloroflexi (1, 10 and 4% for Cape, Angola and Guinea basins, respectively), Planctomycetes (6, 4 and 10%), Acidobacteria (4, 7 and 5%) and Bacteroidetes (10, 4 and 6%).
Bacterial diversity comparison
The proportion of bacteria present in two or three basins was high in the 16S rRNA gene sequences analyses (23%) and in the T-RFLP analyses (58%) (Figure 4a). A third of the fragments (93 TRFs) was detected in the sediments of all basins and represented 82% of the total relative fluorescence intensities. Among the 16S rRNA gene sequences, a shared membership of 19 OTUs (98% identity) was found in all three basins with the statistical tool SONS. The manual assignment in ARB confirmed the small fraction of OTUs detected in all three basins (29 OTUs, 347 sequences), but provided additional information regarding the sequence abundance and identity of each OTU. These were dominated by Gammaproteobacteria (76%; Figure 4b). In this class, the common members were related to marine heterotrophic aerobic and facultative anaerobic microorganisms (Alteromonadaceae and Oceanospirillaceae), photoheterotrophic aerobic bacteria (NOR5/OM60 clade) (Fuchs et al., 2007), and to groups consisting of uncultivated bacteria (JTB255/BD3-6, JTB23/Sva0091/BD3-1, BD7-8/MERTZ, Gret-1F and South Ionian).

(a) Shared and basin-specific SONS generated OTU0.02 and terminal restriction fragment (TRF) and corresponding relative fluorescence intensities (RFI) values for the three deep-sea basin of the South Atlantic Ocean. (b) Phylogeny of the shared OTUs between three and two deep-sea basins and the corresponding number of sequences.
Phylotypes present in two of three basins belonged to the Gamma- and Deltaproteobacteria and to the Chloroflexi. A major group of Chloroflexi-OTUs were restricted to Angola and Guinea basin sediments (7 OTUs, 21 sequences). The ∫-LIBSHUFF analyses revealed no significant difference between the Angola and Guinea libraries as well as Cape and Guinea libraries (using a minimum P-value of 0.0012) (Supplementary Table S1). Thus, common phylotypes dominate the communities of these basins. The largest number of TRFs covered by two basins was found for the Angola and Guinea basins (30 TRFs, 30.5 relative fluorescence intensity) (Figure 4a). High chlorophyll a contents were detected in the Angola and Guinea surface sediments indicating a large fraction of fresh, recently arrived organic carbon (Table 1, Türkay and Kröncke, in preparation). This probably originated from a primary productivity in the surface waters that can be linked to the discharge of nutrients from the Congo and the Niger Rivers into the Angola and Guinea basins, respectively (Schefuss et al., 2004).
Angola and Cape basins showed significantly different communities (∫-LIBSHUFF test, P=0.008) and significantly different Flavobacteria T-RFLP profiles (Figure 5b) (analysis of similarity, R values 0.869, P<0.001) (Supplementary Table S2). These differences were consistent with a different chlorophyll a content as well as a different sediment particle size in the Cape Basin (Table 1) (Etter and Grassle, 1992), indicating that environmental factors seem to influence bacterial communities in deep-sea sediments of the eastern South Atlantic Ocean. It is, however, needed to also take spatial parameters into account in this analysis to strengthen our interpretation concerning environmental or spatial effects on the observed community shifts.

(a) Non-metric multidimensional scaling (nMDS) plot using Bray–Curtis distance for the data set derived from amplification with bacterial primers 27F and 907R and subsequent digestion with AluI. Stress: 0.15. (b) nMDS plot using Bray–Curtis distance for the data set derived from amplification with Flavobacteria-specific primers 517F and 1457R and digestion with MspI. Stress: 0.07.
Biogeography: environmental and historical factors
In the eastern South Atlantic Ocean the Walvis Ridge separates the Cape Basin from the Angola and Guinea basins below a depth of about 3000 m and causes different deep water masses in these basins. The dominance of common phylotypes in the 16S rRNA gene libraries and T-RFLP master profiles suggested that microbial dispersal may not be influenced by the Walvis Ridge or by the presence of different water masses. This was supported by the significantly similar distance–decay relationships of the TRFs in the pairwise comparison (Cape/Angola, slope coefficient 6.9 × 10–5 and 95% confidence interval (3.4 × 10–5, 10.3 × 10–5); Angola/Guinea, slope coefficient 8.7 × 10–5 and 95% confidence interval (1.5 × 10–5, 15.9 × 10–5)). Phylotypes common in the communities of the South Atlantic Ocean and the Pacific, Antarctic and Arctic Oceans sediments (Supplementary Table S1) indicated that some microorganisms disperse effectively over a huge distance and therefore are cosmopolitan, at least at the resolution of 16S rRNA genes that is insufficient for the classification of microorganisms into species (Konstantinidis and Tiedje, 2005).
To get more information regarding the amount of spatial structure present, we analyzed the relative relationships between genetic diversity and geographic distances. The 16S rRNA gene and TRFs based distance–decay relationships for the South Atlantic Ocean and for all sites were all very low (0.003 to 0.07) (Table 3), as also found in taxa-area relationships for soil and salt marsh communities (0.03 to 0.074) (Green et al., 2004; Horner-Devine et al., 2004; Fierer and Jackson, 2006), suggesting high dispersal rates and low extinction rates because of vast population sizes (Connor and McCoy, 1979).
From the clustering of TRF profiles by basins, as shown by non-metric multidimensional scaling (Figure 5a) associated with large, significant R values for all pairwise comparisons between the deep-sea basins (analysis of similarity, 0.586 to 0.999, P<0.001) (Supplementary Table S2), and from significant differences between the South Atlantic Ocean communities to all other communities (∫-LIBSHUFF tests, Supplementary Table S1), it seemed obvious that communities were structured either by the contemporary environment, spatial distances (historical events) or by a combination of both (Martiny et al., 2006; Ramette and Tiedje 2007). To disentangle the relative influence of environmental heterogeneity and spatial distance on the distribution of microbial deep-sea sediment communities, we used a combination of simple and partial Mantel tests. For distances of 0–1200 km T-RFLP results showed a comparable influence of both factors (environment r=0.636, P<0.001, geography r=0.651, P<0.001) (Table 2) (Figure 6b). But environment (r=0.588, P<0.001) overwhelmed any effect of geographic factors (r=0.278, P=0.009) for intermediate distances (1200–3500 km), as also supported by significant partial Mantel tests (Table 3). A higher correlation between spatial and genetic distance for small spatial scales (<200 m) was reported for other microbial groups in soil (Cho and Tiedje, 2000), suggesting the existence of endemic taxa, as genetic distance increases with spatial distances.

Scatterplots with loess curve presenting (a) genetic dissimilarity plotted against geographic distance for genetic dissimilarity of distance matrices derived from 16S ribosomal RNA (rRNA) gene sequences of samples of the South Atlantic and (b) for terminal restriction fragment length polymorphism (T-RFLP) profiles of samples from five different stations in the South Atlantic (Cape, Angola, Guinea I, Guinea II and Guinea III).
Mantel tests for 16S rRNA gene sequences revealed that both environment and geography (r=0.008, P=0.006 and r=0.024, P=0.001, respectively) had an influence on the bacterial diversity of the South Atlantic. Significant correlations between genetic and geographic distances (Mantel's coefficient r=0.013, P=0.001) (Table 3, Figure 6a) were as well observed for all deep-sea and coastal sediments.
When compared in more detail, the two methods showed different results for the relative influence of both factors on microbial biogeography. These detected differences reflect different levels of similarity, saturation and resolution of each method and sampling effort, for example, the T-RFLP analyses including Guinea I versus Guinea I+II+III (Table 3). Indeed, the analysis of 16S rRNA gene sequences provides information regarding randomly chosen phylotypes (‘sampling communities’) where the finding of an OTU is proportional to its abundance in the clone library (Bent and Forney, 2008). In contrast, the fingerprinting method T-RFLP screens for all OTUs present above the detection threshold of the method (‘screening’ communities; Bent and Forney 2008), typically >6 × 102–103 DNA fragment copies per ml samples (Ramette 2009), but does not provide clear taxonomic distinction (Dunbar et al., 2001).
Although high dispersal rates were detected for some groups in deep-sea sediments, both T-RFLP and 16S rRNA-based analyses suggest barriers for the dispersal of microorganisms in the deep sea. The influence of both factors at intermediate scales was already shown by other studies (Green et al., 2004; Reche et al., 2005; Yannarell and Triplett, 2005), but our study suggest an effect of both factors for large scales as well, as shown for soil microbial communities (Fierer and Jackson, 2006). Although the small size, high dispersal rates, large population size and low extinction rates of microorganisms suggest a low effect of geographic barriers on microorganisms (Staley and Gosink, 1999; Beja et al., 2002; Finlay, 2002; Ramette and Tiedje, 2007), our study shows that the distribution of microorganisms in deep-sea sediments is limited at intermediate (10–3000 km) and large scales (>3000 km).
Accession codes
References
Abell GCJ, Bowman JP . (2005a). Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol Ecol 51: 265–277.
Abell GCJ, Bowman JP . (2005b). Colonization and community dynamics of class Flavobacteria on diatom detritus in experimental mesocosms based on Southern Ocean seawater. FEMS Microbiol Ecol 53: 379–391.
Baas-Becking LGM . (1934). Geobiologie of Inleiding Tot De Milieukund. WP Van Stockum and Zoom (in Dutch): The Hague, the Netherlands.
Barnett PRO, Watson J, Connelly D . (1984). A multiple corer for taking virtually undisturbed samples from shelf, bathyal and abyssal sediments. Oceanol Acta 7: 399–408.
Beja O, Koonin EV, Aravind L, Taylor LT, Seitz H, Stein JL et al. (2002). Comparative genomic analysis of archaeal genotypic variants in a single population and in two different oceanic provinces. Appl Environ Microbiol 68: 335–345.
Bent SJ, Forney LJ . (2008). The tragedy of the uncommon: understanding limitations in the analysis of microbial diversity. ISME J 2: 689–695.
Bickert T, Wefer G . (1996). Late quaternary deep water circulation in the South Atlantic: reconstruction from carbonate dissolution and benthic stable isotopes. In: Berger WH, Wefer G, Siedler G (eds). The South Atlantic: Present and Past Circulation. Springer: Heidelberg, pp 599–620.
Bowman JP, McCuaig RD . (2003). Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl Environ Microbiol 69: 2463–2483.
Brandt A, Brenke N, Andres HG, Brix S, Guerrero-Kommritz E, Muhlenhardt-Slegel U et al. (2005). Diversity of peracarid crustaceans (Malacostraca) from the abyssal plain of the Angola Basin. Org Divers Evol 5: 105–112.
Cho JC, Tiedje JM . (2000). Biogeography and degree of endemicity of fluorescent Pseudomonas strains in soil. Appl Environ Microbiol 66: 5448–5456.
Connor EF, McCoy ED . (1979). Statistics and biology of the species-area relationship. Amer Nat 113: 791–833.
Deming JW, Colwell RR . (1982). Barophilic bacteria associated with digestive tracts of abyssal holothurians. Appl Environ Microbiol 44: 1222–1230.
Dunbar J, Ticknor LO, Kuske CR . (2001). Phylogenetic specificity and reproducibility and new method for analysis of terminal restriction fragment profiles of 16S rRNA genes from bacterial communities. Appl Environ Microbiol 67: 190–197.
Etter RJ, Grassle JF . (1992). Patterns of species-diversity in the deep-sea as a function of sediment particle-size diversity. Nature 360: 576–578.
Fierer N, Jackson RB . (2006). The diversity and biogeography of soil bacterial communities. Proc Natl Acad Sci USA 103: 626–631.
Finlay BJ . (2002). Global dispersal of free-living microbial eukaryote species. Science 296: 1061–1063.
Fuchs BM, Spring S, Teeling H, Quast C, Wulf J, Schattenhofer M et al. (2007). Characterization of a marine gammaproteobacterium capable of aerobic anoxygenic photosynthesis. Proc Natl Acad Sci USA 104: 2891–2896.
Grant A . (2000). Deep-sea diversity: overlooked messages from shallow-water sediments. Mar Ecol 21: 97–112.
Green JL, Holmes AJ, Westoby M, Oliver I, Briscoe D, Dangerfield M et al. (2004). Spatial scaling of microbial eukaryote diversity. Nature 432: 747–750.
Guezennec J, Fiala-Medioni A . (1996). Bacterial abundance and diversity in the Barbados Trench determined by phospholipid analysis. FEMS Microbiol Ecol 19: 83–93.
Harvey HR, Richardson MD, Patton JS . (1984). Lipid-composition and vertical-distribution of bacteria in aerobic sediments of the Venezuela basin. Deep Sea Res Part A 31: 403–413.
Hewson I, Fuhrman JA . (2006). Improved strategy for comparing microbial assemblage fingerprints. Microb Ecol 51: 147–153.
Horner-Devine MC, Lage M, Hughes JB, Bohannan BJM . (2004). A taxa-area relationship for bacteria. Nature 432: 750–753.
Huber T, Faulkner G, Hugenholtz P . (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20: 2317–2319.
Hughes DJ, Gage JD . (2004). Benthic metazoan biomass, community structure and bioturbation at three contrasting deep-water sites on the northwest European continental margin. Prog Oceanogr 63: 29–55.
Kato C, Yanagibayashi M, Nogi Y, Li L, Horikoshi K . (1999). Changes in the microbial community in deep-sea sediment during cultivation without decompression. In: Ludwig H (ed). Advances in High Pressure Bioscience and Biotechnology. Springer: Heidelberg, pp 11–15.
Konstantinidis KT, Tiedje JM . (2005). Towards a genome-based taxonomy for prokaryotes. J Bacteriol 187: 6258–6264.
Legendre P, Legendre L . (1998). Numerical Ecology. Elsevier Science BV: Amsterdam.
Li L, Kato C, Horikoshi K, Tsujii K . (1999). Microbial diversity in the sediments collected from cold-seep areas and from different depths of the deep-sea. In: Horikoshi K, Tsujii K (eds). Extremophiles in Deep-Sea Environments. Springer: Tokyo, pp 55–88.
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res 32: 1363–1371.
Maidak BL, Olsen GJ, Larsen N, Overbeek R, McCaughey MJ, Woese CR . (1996). The Ribosomal Database Project (RDP). Nucleic Acids Res 24: 82–85.
Mantel N . (1967). Detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4: 102–112.
Muyzer G, Teske A, Wirsen CO, Jannasch HW . (1995). Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel-electrophoresis of 16S rDNA fragments. Arch Microbiol 164: 165–172.
O’Malley MA . (2008). ‘Everything is everywhere: but the environment selects’: ubiquitous distribution and ecological determinism in microbial biogeography. Stud Hist Philos Biol Biomed Sci 39: 314–325.
Osborn AM, Moore ERB, Timmis KN . (2000). An evaluation of terminal-restriction fragment length polymorphism (T-RFLP) analysis for the study of microbial community structure and dynamics. Environ Microbiol 2: 39–50.
Papke RT, Ramsing NB, Bateson MM, Ward DM . (2003). Geographical isolation in hot spring cyanobacteria. Environ Microbiol 5: 650–659.
Polymenakou PN, Bertilsson S, Tselepides A, Stephanou EG . (2005). Bacterial community composition in different sediments from the Eastern Mediterranean Sea: a comparison of four 16S ribosomal DNA clone libraries. Microb Ecol 50: 447–462.
Polymenakou PN, Lampadariou N, Mandalakis M, Tselepides A . (2009). Phylogenetic diversity of sediment bacteria from the southern Cretan margin, Eastern Mediterranean Sea. Syst Appl Microbiol 32: 17–26.
Queric NV, Soltwedel T . (2007). Impact of small-scale biogenic sediment structures on bacterial distribution and activity in Arctic deep-sea sediments. Mar Ecol-Evol Persp 28: 66–74.
Ramette A . (2009). Quantitative community fingerprinting methods for estimating the abundance of operational taxonomic units in natural microbial communities. Appl Environ Microbiol 75: 2495–2505.
Ramette A, Tiedje JM . (2007). Biogeography: an emerging cornerstone for understanding prokaryotic diversity, ecology, and evolution. Microb Ecol 53: 197–207.
Ravenschlag K, Sahm K, Pernthaler J, Amann R . (1999). High bacterial diversity in permanently cold marine sediments. Appl Environ Microbiol 65: 3982–3989.
Reche I, Pulido-Villena E, Morales-Baquero R, Casamayor EO . (2005). Does ecosystem size determine aquatic bacterial richness? Ecology 86: 1715–1722.
Rossello-Mora R, Amann R . (2001). The species concept for prokaryotes. FEMS Microbiol Rev 25: 39–67.
Santelli CM, Orcutt BN, Banning E, Bach W, Moyer CL, Sogin ML et al. (2008). Abundance and diversity of microbial life in ocean crust. Nature 453: 653–U657.
Schefuss E, Versteegh GJM, Jansen JHF, Damste JSS . (2004). Lipid biomarkers as major source and preservation indicators in SE Atlantic surface sediments. Deep-Sea Res Pt I 51: 1199–1228.
Schloss PD, Handelsman J . (2005). Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71: 1501–1506.
Schloss PD, Handelsman J . (2006). Introducing SONS, a tool for operational taxonomic unit-based comparisons of microbial community memberships and structures. Appl Environ Microbiol 72: 6773–6779.
Schloss PD, Larget BR, Handelsman J . (2004). Integration of microbial ecology and statistics: a test to compare gene libraries. Appl Environ Microbiol 70: 5485–5492.
Shannon LV, Chapman P . (1991). Evidence of Antarctic bottom water in the Angola Basin at 32-degrees-S. Deep Sea Res A 38: 1299–1304.
Stackebrandt E, Ebers J . (2006). Taxonomic parameters revisited: tarnished gold standards. Microbiol Today 33: 152–155.
Staley JT, Gosink JJ . (1999). Poles apart: biodiversity and biogeography of sea ice bacteria. Annu Rev Microbiol 53: 189–215.
Suzuki M, Rappe MS, Giovannoni SJ . (1998). Kinetic bias in estimates of coastal picoplankton community structure obtained by measurements of small-subunit rRNA gene PCR amplicon length heterogeneity. Appl Environ Microbiol 64: 4522–4529.
Urakawa H, Kita-Tsukamoto K, Ohwada K . (1999). Microbial diversity in marine sediments from Sagami Bay and Tokyo Bay, Japan, as determined by 16S rRNA gene analysis. Microbiology-(UK) 145: 3305–3315.
Whitaker RJ, Grogan DW, Taylor JW . (2003). Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science 301: 976–978.
Xu MX, Wang P, Wang FP, Xiao X . (2005). Microbial diversity at a deep-sea station of the Pacific nodule province. Biodivers Conserv 14: 3363–3380.
Yan S, Fuchs BM, Lenk S, Harder J, Wulf J, Jiao NZ et al. (2009). Biogeography and phylogeny of the NOR5/OM60 clade of Gammaproteobacteria. Syst Appl Microbiol 32: 124–139.
Yannarell AC, Triplett EW . (2005). Geographic and environmental sources of variation in lake bacterial community composition. Appl Environ Microbiol 71: 227–239.
Zhao J, Zeng RY . (2005). Phylogenetic analysis of bacterial community in deep-sea sediment from the western Pacific ‘warm pool’. Acta Oceanol Sin 24: 127–138.
Zheng DD, Alm EW, Stahl DA, Raskin L . (1996). Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl Environ Microbiol 62: 4504–4513.
Zhou JZ, Xia BC, Treves DS, Wu LY, Marsh TL, O’Neill RV et al. (2002). Spatial and resource factors influencing high microbial diversity in soil. Appl Environ Microbiol 68: 326–334.
Acknowledgements
We thank M Türkay and P Martínez Arbizu for the invitation to participate to the Meteor cruise DIVA II (M63/2) and the captain, crew and shipboard scientific party for their assistance during sampling. We are grateful to I Kröncke and M Türkay for providing environmental data of the deep-sea sediments. The project has been carried out in the framework of the Consensus of Diversity of Abyssal Marine Life (CoML/CeDAMar) and the MarBEF Network of Excellence ‘Marine Biodiversity and Ecosystem Functioning’ (contract no. GOCE-CT-2003-505446). This work was financially supported by the Max Planck Society.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Rights and permissions
About this article
Cite this article
Schauer, R., Bienhold, C., Ramette, A. et al. Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J 4, 159–170 (2010). https://doi.org/10.1038/ismej.2009.106
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2009.106
Keywords
This article is cited by
-
Microbe-driven elemental cycling enables microbial adaptation to deep-sea ferromanganese nodule sediment fields
Microbiome (2023)
-
Microbial communities in the deep-sea sediments of the South São Paulo Plateau, Southwestern Atlantic Ocean
International Microbiology (2023)
-
Environmental and Spatial Influences on Biogeography and Community Structure of Saltmarsh Benthic Diatoms
Estuaries and Coasts (2021)
-
Microbial community and geochemical analyses of trans-trench sediments for understanding the roles of hadal environments
The ISME Journal (2020)
-
Evidence for phylogenetically and catabolically diverse active diazotrophs in deep-sea sediment
The ISME Journal (2020)
