
Abstract
Microbial ecology is a field that applies molecular techniques to analyze genes and communities associated with a plethora of unique environments on this planet. In the past, low biomass and the predominance of a few abundant community members have impeded the application of techniques such as PCR, microarray analysis and metagenomics to complex microbial populations. In the absence of suitable cultivation methods, it was not possible to obtain DNA samples from individual microorganisms. Recently, a method called multiple displacement amplification (MDA) has been used to circumvent these limitations by amplifying DNA from microbial communities in low-biomass environments, individual cells from uncultivated microbial species and active organisms obtained through stable isotope probing incubations. This review describes the development and applications of MDA, discusses its strengths and limitations and highlights the impact of MDA on the field of microbial ecology. Whole genome amplification via MDA has increased access to the genomic DNA of uncultivated microorganisms and low-biomass environments and represents a ‘power tool’ in the molecular toolbox of microbial ecologists.
Similar content being viewed by others


Introduction
Environmental microbial diversity is poorly understood and the majority of microbes are inaccessible by laboratory cultivation. As a result, microbial ecology has sought to develop molecular methods to characterize whole communities. The extraction of community nucleic acids is a typical initial step, and although cell biomass is often sufficiently high to enable analysis by PCR, gene hybridization or metagenomics, this is not always the case. Because microbial ecologists study challenging environments, such as insect guts (Broderick et al., 2004), ice cores (Christner et al., 2001), permafrost (Steven et al., 2006), deep subsurface sediments (Teske, 2005) and air (Brodie et al., 2007), high-sensitivity PCR protocols have been required for the analysis of single genes. A whole genome amplification (WGA) step provides access to community DNA from these low-biomass environments. In addition, the vast number of uncultivated organisms associated with the ‘rare biosphere’ (Sogin et al., 2006) contain enzyme-encoding genes that hold great promise for medicine, biotechnology and industry. Fortunately for microbial ecologists in the early 21st century, small yields of DNA from low-biomass communities or individual uncultivated cells are readily retrieved by the advent of WGA via the multiple displacement amplification (MDA) reaction. Here we highlight the history of MDA and discuss its mechanism, capabilities and limitations. The applications of MDA in microbial ecology are reviewed for accessing low-abundance DNA from cells or environmental samples. Finally, we discuss the future of MDA and its potential integration into additional facets of microbial ecology research.
A brief history of MDA
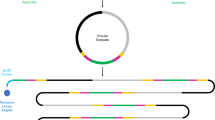
Initial WGA reactions utilized PCR-based techniques such as degenerate oligonucleotide primed PCR (Telenius et al., 1992) and primer extension PCR (Zhang et al., 1992). However, these were limited by nonspecific artifacts of amplification (Cheung and Nelson, 1996), strong bias (Paunio et al., 1996) and short amplification products (Telenius et al., 1992; Zhang et al., 1992; Paunio et al., 1996). MDA was the first WGA method based on an isothermal reaction (Dean et al., 2001). Double-stranded DNA template is initially denatured before incubating at 30 °C for 2–16 h, depending on the amount of starting template and the commercial MDA kit being used (Table 1). MDA utilizes target DNA template, buffer, dNTPs, random phosphorothioate-modified hexamers and DNA polymerase, which is commonly derived from the Bacillus subtilis bacteriophage phi29. The 3′ phosphorothioate modifications are required for amplification, as they render the hexamers resistant to the 3′–5′ exonuclease proofreading activity of the polymerase (Dean et al., 2001). The phi29 DNA polymerase extends the random primers and its strong ‘strand displacement activity’ allows it to displace existing primer-originated extensions downstream (Figure 1). Continued priming and strand displacement generates a branched structure and gives exponential DNA amplification. The reaction yields double-stranded linear DNA, single-stranded forms and some remaining branched intermediate structures (Figure 1). Complete denaturation of MDA products and subsequent resolution on denaturing alkaline agarose gels revealed a 12 kb average length of the resulting single-stranded amplified DNA (Dean et al., 2002). The reaction is terminated by heating to 65 °C for 10 min, denaturing the polymerase, and yields up to 40 μg of DNA per 50-μl reaction.

Sequential diagrammatic representation of branched structures formed during multiple displacement amplification (MDA). The arrowheads represent the location of the phi29 polymerase synthesizing DNA in a 5′ to 3′ direction.
The phi29 DNA polymerase was first isolated by Blanco and Salas (1984) from Escherichia coli cells expressing the P2 gene, which encodes the sole phi29-associated DNA polymerase. It has an extremely high processivity, adding an average of 70 000 nucleotides each time it binds the primer template (Blanco et al., 1989). Use of single-stranded M13 DNA as template gave a rolling-circle mode of replication, in which the polymerase repeatedly copied around the circular template via its strand displacement activity, yielding a product of concatenated M13 repeats. The DNA polymerase's associated 3′–5′ exonuclease proofreading activity results in a low intrinsic error rate of 10−6–10−7 (Watabe et al., 1984; Blanco and Salas, 1985) and the accumulation of mutations in MDA products at a rate of only 10−5–10−6 (Esteban et al., 1993; Nelson et al., 2002), nearly 1000-fold less than for PCR using the Taq DNA polymerase (Dunning et al., 1988; Saiki et al., 1988).
The first use of MDA for amplifying whole genomes targeted human DNA (Dean et al., 2002) and as few as 90 copies of the genome (0.3 ng DNA) yielded more than 30 μg of product. For eight loci assessed, amplification bias (over- and underrepresentation of sequences) was several orders of magnitude less than for the PCR-based WGA (Dean et al., 2002). Since then, MDA has been applied to many more eukaryotic studies, including the genomic sequencing and genotyping of humans and primates (Lovmar et al., 2003; Barker et al., 2004; Paez et al., 2004; Dickson et al., 2005; Jiang et al., 2005; Lu et al., 2005; Rönn et al., 2006), insects (Gorrochotegui-Escalante and Black, 2003), fungi (Foster and Monahan, 2005; Gadkar and Rillig, 2005; Fernández-Ortuño et al., 2007) and additional applications in forensics and medicine (Hosono et al., 2003; Lasken and Egholm, 2003; Barber and Foran, 2006; Spits et al., 2006; Ballantyne et al., 2007).
Although MDA has enabled genomic sequencing from low concentrations of template nucleic acid, remaining limitations include (a) nonspecific amplification derived from primer dimer formation or contaminating DNA template, (b) formation of chimeric DNA rearrangements and (c) representation bias. Exciting new research indicates the possibility of improvements to the specificity, fidelity and sensitivity of MDA. For example, single-strand binding protein and spermidine were reported to improve DNA yield up to 66%, with reduced representational bias and increased sensitivity for targeted templates (Wu et al., 2006). Similar effects were reported with the mutant single-strand binding protein protein of Thermus thermophilus (Inoue et al., 2006). One report suggested that background synthesis from primer–primer interactions may also be reduced or eliminated by the modification of hexamer primers with the incorporation of one or two 5′-terminal universal bases (Lage et al., 2003). Recent work demonstrated that carefully avoiding trace levels of DNA contamination resulted in only 0.36–1.0% of sequences being nonspecific (Marcy et al., 2007a).
The concentration and copy number of DNA template affect the MDA reaction. Hutchison et al. (2005) discovered that a smaller MDA reaction volume (3 μl) greatly reduced the amount of nonspecific DNA generated with 50 molecules of single-stranded circular M13 template compared to higher reaction volumes (15 or 30 μl). Others have shown that the total number of DNA templates affects representational bias (Lasken et al., 2005), with a single copy of template giving the highest bias (Raghunathan et al., 2004). Reducing bacterial DNA template from 10 ng (>106 genome copies) by 10- and 100–1000-fold reduced representational bias 3- and 6-fold, respectively (Wu et al., 2006). Other studies also showed increased bias with decreased template copy number (Detter et al., 2002; Bergen et al., 2005; Holbrook et al., 2005; Kalyuzhnaya et al., 2006; Neufeld et al., 2008).
Chimera formation and amplification bias are concerns for genomic sequencing of DNA generated from MDA. Zhang et al. (2006) discovered that cloned MDA products included up to 50% chimeric inserts (3 kb average length). They postulated (incorrectly) that chimeras were generated by the cloning step, with E. coli rearranging the cloned and branched DNA. Remarkably, chimeras were reduced by 80% (to a final rate of 6% of inserts screened) through three enzymatic treatments of (a) phi29 DNA polymerase and dNTPs in the absence of primers to ‘debranch’ the DNA, (b) S1 nuclease to digest remaining single-stranded DNA present in the mixture and (c) flow shearing and size selection on agarose gels followed by nick translation with E. coli DNA polymerase I.
The predominant mechanism for chimera formation has now been elucidated and occurs during the MDA reaction rather than in the subsequent cloning step (Lasken and Stockwell, 2007). Pyrosequencing (454 Life Sciences method; a cloning-independent approach) of DNA amplified from single E. coli cells revealed a chimera frequency (one chimeric junction per 22 kb) similar to that previously detected (Zhang et al., 2006) by cloning-dependent Sanger sequencing. The principal mechanism of chimera formation was through initial extension of random primers followed by displacement of these 3′ ends and re-priming on a second template, often where there was a short region of complimentarity of 2–21 bp. The second templates were usually nearby 5′ ends such that 85% of chimeras consisted of two segments joined in inverted orientation (Lasken and Stockwell, 2007). It was suggested that after amplification, digestion with S1 nuclease (Zhang et al., 2006) may be effective by digesting single-stranded regions postulated to occur as MDA reaction intermediates in the pathway leading to chimeras (Lasken and Stockwell, 2007).
Use of MDA in microbial ecology
Single-cell amplification
Microbial ecology is in the early stages of establishing links between uncultivated microorganisms in the environment and their trophic roles within communities (Neufeld et al., 2007). Progress has been made in the ability to analyze genomes, without the prerequisite of cultivation, by using MDA from individual cells. The first demonstration of genomic sequencing from single cells used E. coli isolated by fluorescence-activated cell sorting, followed by MDA (Raghunathan et al., 2004). Amplification of ∼5 fg of template generated ∼24 μg of product, more than a billion-fold amplification. No miscalls were found in a 662-bp 16S rRNA gene sequence amplified from MDA DNA, demonstrating that cultivation was no longer a prerequisite for genomic analysis. Owing to nonspecific DNA synthesis, only 30% of the amplified DNA was actually E. coli genomic sequence. Recent work has achieved ⩾99% specific amplification from a single cell (Marcy et al., 2007a), possibly due to more stringent protocols for avoiding trace DNA contaminants. MDA was first used to sequence from an uncultured cell isolated directly from the environment using fluorescence in situ hybridization (FISH) of 16S rRNA to identify and capture soil microbes by micromanipulation (Lasken et al., 2005). Micromanipulation is relatively time consuming but is a powerful research tool allowing observation of cell morphology, repeated washing in rinse buffers to remove contaminating free DNA and visual documentation that a single cell was captured and delivered to the MDA reaction (Ishøy et al., 2006; Kvist et al., 2007). As discussed below, the isolation of single or a few cells for use in MDA reactions has also been carried out by fluorescence-activated cell sorting (Raghunathan et al., 2004; Podar et al., 2007; Stepanauskas and Sieracki, 2007), microfluidics (Marcy et al., 2007a, 2007b) or micromanipulation (Kvist et al., 2007). Cell sorting by FISH labelling of 16S rRNA followed by automated cell lysis and MDA using liquid-handling stations is expected to provide unprecedented high throughput for the analysis of uncultivated cells.
Single-cell analysis is complementary to metagenomics (Lasken, 2007), which uses bulk environmental DNA samples to rapidly obtain genomic sequence but can be limited by the difficulty of assembling individual genomes from multiple organisms (Tringe et al., 2005; Rusch et al., 2007). Even partial genomic drafts from single cells provide genetically linked sequences useful for guiding the assembly of metagenomic sequences into discrete genomes (Lasken, 2007). In a proof of principle for community analysis, cells from a defined mixture of three methylotrophic bacteria were separated by flow sorting (Kalyuzhnaya et al., 2006) using 16S rRNA FISH probes that distinguished between these three methylotrophs. When 101, 102 and 103 cells were collected in each tube and subjected to MDA, 20%, 50% and 95% of resulting DNA sequences correctly associated with the FISH-targeted organism, and none were from the ‘contaminating’ organisms; the remaining sequences corresponded to nonspecific DNA synthesis generated as an artifact of MDA. All of the tested clones represented the desired microbe when 104 and 105 cells were used as template in MDA.
For environmental samples, the use of FISH probes for 16S rRNA in fluorescence-activated cell sorting prior to MDA was demonstrated by Podar et al. (2007) targeting the uncultivated phylum TM7, which existed at low relative abundance (⩽2%) in a California soil sample. Amplification of DNA from five sorted cells resulted in detection of the TM7 16S rRNA gene, but also labelled sequences from contaminating Pseudomonas spp DNA. Nonetheless, they collected >20 000 sequences that assembled into 132 contigs, encoding 670 predicted TM7 proteins. The TM7 representatives were found to share similarity to Chloroflexi and possess genes associated with multidrug resistance, starvation resistance and DNA restriction modification. Further, TM7 cells possibly employ a twitching motility and may be involved in biofilm formation, both suggestions supported by the retrieval of genes encoding a Type IV pilus. Organisms from the TM7 phylum also occur at a low relative abundance (⩽2%) on human subgingival tooth surfaces. Single TM7 cells were examined in dental plaque communities with a FISH-targeted, custom-designed microfluidic system coupled with two rounds of MDA (Marcy et al., 2007b). Of the 2.86 Mb sequence data obtained from cloned DNA, 1474 genes were identified across 288 scaffolds with ⩾3 genes. Several genes were associated with Type IV pilus sequences, similar to those found in soil TM7 cells (Podar et al., 2007). Together, these studies offered a previously unavailable glimpse into the potential metabolism and community function of this widespread and uncultivated bacterial division.
In another study (Kvist et al., 2007), micromanipulation was used to retrieve the uncultivated clade C1b crenarchaeotes from agricultural soil in Denmark. These may be major contributors to ammonia oxidation in world soils (Leininger et al., 2006), yet they have only been detected in agricultural soil clone libraries (Ochsenreiter et al., 2003), a fosmid metagenomic library from a soil sample (Quaiser et al., 2002) and in root-extract enrichment cultures (Simon et al., 2005). Cells were disrupted from the soil matrix, isolated with a micromanipulation method for individual FISH probe-positive cells (Ishøy et al., 2006) and used for MDA and DNA sequencing (Kvist et al., 2007). Clone libraries confirmed that DNA from C1b phylotypes was amplified for two of the eight collected cells. Sequencing 97 plasmid clones yielded 130 kb and more than 60 open reading frames, 20 of which were unambiguously associated with the collected crenarchaeote. The remaining clones were of unknown origin, but were quite possibly associated with the archaeal cells collected in this study. Notably, one of the open reading frames closely matched a nitrous oxide reductase from an uncultivated soil bacterium, indicating that some soil crenarchaeotes may be capable of dissimilatory nitrous oxide reduction.
As an alternative to sorting cells with fluorescence, cells or extracted genomic DNA from particular organisms may be amenable to alternative purification methods prior to MDA. Mavingui et al. (2005) purified cells and DNA of an obligate intracellular pathogen (Wolbachia pipientis) from a large amount of host material, using differential centrifugation and pulsed-field gel electrophoresis. The ∼50 ng of retrieved Wolbachia DNA was amplified via MDA. Multiple PCR primer sets confirmed that the MDA products preserved the gene arrangements found in the original template. In another study, individual filaments, each containing several hundred cells of the large sulfur bacteria Beggiatoa, were physically removed from contaminating sediment cells and subjected to MDA (Mußmann et al., 2007). Sequences representing an estimated 70% of the genome were then obtained with 454 pyrosequencing. The subsequent bioinformatic analysis confirmed the chemolithoautotrophic metabolism proposed for Beggiatoa in 1888, and suggested that other electron donors and acceptors may be coupled in this fascinating sulfur-oxidizing bacterium.
Methods for isolating cells and characterizing their genomes are expected to become increasingly sophisticated and high throughput (Lasken, 2007; Marcy et al., 2007a), especially methods that target organisms associated with the ‘rare biosphere’ (Sogin et al., 2006) and techniques specifically linked to the incorporation of labelled substrates (Huang et al., 2007). As a result, the use of MDA to obtain genomic DNA from single cells is anticipated to play a central and increasing role in accessing genetic and ecological information associated with uncultivated microorganisms.
Whole community genome amplification
Microbial ecologists have used DNA-based methodologies to study the diversity and composition of microbial communities in biomass-rich environments for the past two decades. However, the study of low-biomass environments with PCR, DNA hybridization (for example, microarrays and dot-blot hybridization) or metagenomics has been limited by contaminating humic acids (for PCR) and low yields of extracted nucleic acid for community analyses (for PCR, hybridization and metagenomics). Metagenomics provides a broad view of a community's genetic composition, but requires microgram quantities of DNA to construct libraries for screening or sequencing. While extracted DNA may not be limiting from surface soils or sediments, it may be limiting from environments of low bacterial abundance. Whole community genome amplification using MDA has recently overcome these limitations, enabling the molecular assessment of microbial communities that would not have been accessible otherwise.
One difficulty for low-biomass environments is that associated humic acids and exopolysaccharides may inhibit PCR-based analysis. To alleviate this, Gonzalez et al. (2005) used MDA as a pre-PCR enrichment step on samples from a cave, meadow soil and a wastewater treatment system. MDA from a defined mixture of pure-culture DNA accurately reflected the unamplified mixture based on denaturing gradient gel electrophoresis fingerprints. An analysis of Wolbachia DNA from infected mites (Jeyaprakash and Hoy, 2004) and a study of Mycobacterium leprae from human skin samples (Groathouse et al., 2006) have also demonstrated MDA as a powerful preamplification step when PCR is hindered by low template concentration or coextracted cellular PCR inhibitors.
In an example of a community analysis using MDA, microorganisms associated with a lace coral (Pocillopora damicornis) were studied (Yokouchi et al., 2006). Limited sample sizes and the difficulty of extracting quantifiable DNA from coral made this an ideal community to access through MDA. First, MDA representational bias was assessed using cyanobacterium Synechocystis sp PCC6803. Ten single-copy genes assayed by quantitative PCR (after MDA) were all amplified between 104 and 105 times by MDA, a substantial improvement over degenerate oligonucleotide primed PCR and primer extension PCR, which had amplification bias over a range of 3–6 orders of magnitude for tested sequences (Dean et al., 2002). To analyze the coral microbial community, 0.4 ng of extracted DNA was subjected to MDA, yielding 24 μg of ⩾10-kb product. Approximately 70% of sequences were in common between PCR-generated 16S rRNA gene libraries derived from MDA and control (unamplified) DNA, indicating that MDA maintains high representation of community composition. The study demonstrated that Proteobacteria (Alphaproteobacteria in particular) were dominant phylotypes in this coral DNA extract.
Subsurface and contaminated soil microbial populations are examples of communities in which metagenomic library construction would be difficult. Abulencia et al. (2006) estimated that an untenable 11–88 kg of contaminated subsurface soil (at 104 cells g–1) would be necessary to extract microgram amounts of DNA for shotgun library construction, with 20-fold more required for BAC libraries. To overcome this, MDA was successfully applied to heavily contaminated subsurface-soil samples from Oak Ridge, Tennessee, to generate sufficient template for 16S rRNA gene PCR and construction of libraries. Prior to MDA, only one of three samples gave useful libraries. Diversity estimates also indicated that the libraries from amplified DNA extracts possessed a higher level of diversity. Small-insert metagenomic libraries generated from three MDA reactions shared a similar cluster of orthologous group profiles, indicating conservation of sequences by MDA. However, MDA from several microbes, experimentally mixed in known ratios, indicated that some bias may occur in their representation (Abulencia et al., 2006). In another subsurface study, Edwards et al. (2006) demonstrated the first use of 454 pyrosequencing to characterize metagenomic DNA, employing MDA to obtain sufficient template using microbial assemblages from the Soudan Iron Mine in Minnesota, USA. Pyrosequencing of MDA-amplified microbial DNA also shed light on the metagenomic composition of a Caribbean coral holobiont (Wegley et al., 2007). These studies demonstrate the utility of MDA for generation of metagenomic libraries when DNA quantities are insufficient for direct cloning or pyrosequencing.
Viral particles, typically purified by density gradient ultracentrifugation, have also been subjected to MDA to overcome low DNA yields for identification of novel DNA viruses in blood (Breitbart and Rohwer, 2005) and characterization of marine viral assemblages in world oceans (Angly et al., 2006). Viral assemblages of the Sargasso Sea, Gulf of Mexico, coast of British Columbia and the Arctic ocean were compared with viral metagenomic DNA (‘viromes’) from MDA amplifications. In this study, 454 pyrosequencing generated enormous libraries of phage sequences from nanogram quantities of DNA.
Microarrays are high-throughput tools for the functional or phylogenetic assessment of microbial communities, and may also be limited by the amount of DNA in low-biomass environments. Vora et al. (2004) used MDA and microarrays for the detection of enterohemorrhagic E. coli in environmental samples. Low bias and high sensitivity were obtained starting from either pure cultures or spiked environmental samples, demonstrating that this approach would be feasible for pathogen detection and typing. For the microarray analysis of whole communities, MDA was applied to DNA extracted from contaminated groundwater from Oak Ridge, Tennessee (Wu et al., 2006). Dilutions (ranging from 500 ng to 10 pg) of extracted DNA from ethanol-amended sites were used to assess the representational bias associated with MDA. For all starting template quantities above 1 ng, the percentage of genes detected after MDA exceeded 93% of those detected in undiluted DNA. At 0.1 or 0.01 ng of template, only 64% and 51% of detected genes were reflected in the unamplified starting DNA, respectively. Predictably, representational bias was highest for these low DNA template MDA reactions. MDA was also used to amplify DNA from unamended groundwater samples of low biomass (∼104 cells ml−1) that only yielded extracted DNA in the nanogram range. Over 400 genes were detected and indicated that contamination strongly influenced the genetic diversity and composition of the community. The inclusion of an MDA step enabled this soil comparison and alleviated the limitation of sample size for analyzing the microorganisms and genes involved in attenuating pollutant contamination.
Polymerase chain reaction, metagenomics and microarrays enable microbial ecologists to link environmental variables with particular genes and phylotypes, but do not directly implicate particular uncultivated microorganisms with their preferred carbon sources. Stable isotope probing (SIP) is a powerful method that establishes this link between function and phylogeny (Neufeld et al., 2007); however, it has been limited by an experimental ‘catch-22’. Does one add an excess of labelled substrate to an SIP incubation to increase the yield of labelled DNA and potentially bias the experiment toward copiotrophic species capable of rapid cell division? Or does one instead use near-in situ incubation conditions but greatly reduce the yield of labelled nucleic acid? A recent study has overcome this limitation by using in situ concentrations of substrate in a marine SIP incubation and then amplifying the low-nanogram quantities of labelled DNA with MDA prior to metagenomic analysis with a fosmid library (Neufeld et al., 2008). Using 1 μM 13C-methanol for marine surface seawater SIP incubations, the total labelled DNA retrieved from this sample was less than 25 ng, over three orders of magnitude less than the amount required for large-insert metagenomic library construction. MDA generated microgram yields of DNA and 16S rRNA gene fingerprints confirmed that the amplified DNA was highly representative of the original unamplified DNA. This approach led to the retrieval of a methanol dehydrogenase-containing operon of an active marine methylotroph. The metagenomic examination of an active methylotroph population enriched with in situ conditions would not have been feasible without MDA.
In a conceptually similar study, Leigh et al. (2007) used MDA to increase the concentration of DNA that was labelled in a 13C-biphenyl SIP incubation. MDA products were hybridized to the GeoChip, a microarray with probes targeting genes involved in aromatic hydrocarbon degradation (Leigh et al., 2007). One nanogram of DNA was amplified for microarray analysis, further underscoring the usefulness of MDA for studying the microbial ecology of low-biomass environments.
Conclusion
The use of MDA for single-cell DNA amplification and for low concentrations of environmental DNA has only just begun. However, its widespread applicability has quickly become apparent and has allowed microbial ecologists to circumvent several limitations impeding research on the genetics and diversity of uncultivated microbial cells and low-biomass environments. In only three years, MDA has been applied to studies using PCR, cell sorting, microarrays, stable-isotope probing, gel fingerprinting and metagenomics. The integration of MDA into microbial ecology has been encouraged by results indicating that sensitivity is high, representational bias is relatively low (especially for amplifications of ⩾1 ng template) and misincorporated bases are infrequent. The presence of chimeras introduced during MDA is a current drawback that may limit the use of large insert DNA libraries for genomic sequencing. Neufeld et al. (2008) used intergenic PCR to identify chimeras in 10-kb inserts in a metagenomic library generated by MDA, but this labor-intensive step would not be feasible as a means to screen for usable clones in larger-scale projects. However, small-insert libraries worked well for genome assembly (Zhang et al., 2006) when sufficient sequencing depth enabled subtraction of rare chimeras from the multiple reads of the correct sequence. Novel DNA-sequencing methods using shorter DNA read length should result in even fewer chimeric junctions per read as demonstrated for single cells by the 454 Life Sciences method (Marcy et al., 2007a). Nevertheless, the appearance of MDA-associated chimeras in databases of environmental DNA sequences is a concern, as has already occurred for the PCR method in the case of ribosomal sequence databases (Hugenholtz and Huber, 2003; Ashelford et al., 2005). It will be important to reduce the formation of chimeras further, and identifying their cause (Lasken and Stockwell, 2007) is a substantial step in this direction.
While efforts continue to improve the accuracy of MDA, it has already provided microbial ecologists with a tool for increasing the sensitivity of microbial community analysis and has enabled the genetic studies of single cells, both cultivated and uncultivated. MDA is a recent and welcome addition to the toolbox of microbial ecologists.
References
Abulencia CB, Wyborski DL, Garcia JA, Podar M, Chen W, Chang SH et al. (2006). Environmental whole-genome amplification to access microbial populations in contaminated sediments. Appl Environ Microbiol 72: 3291–3301.
Angly FE, Felts B, Breitbart M, Salamon P, Edwards RA, Carlson C et al. (2006). The marine viromes of four oceanic regions. PLoS Biol 4: e368.
Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ . (2005). At least 1 in 20 16S rRNA sequence records currently held in public repositories is estimated to contain substantial anomalies. Appl Environ Microbiol 71: 7724–7736.
Ballantyne K, van Oorschot R, Mitchell R . (2007). Increasing amplification success of forensic DNA samples using multiple displacement amplification. Forensic Science, Medicine, and Pathology 3: 182–187.
Barber AL, Foran DR . (2006). The utility of whole genome amplification for typing compromised forensic samples. J Forensic Sci 51: 1344–1349.
Barker DL, Hansen MST, Faruqi AF, Giannola D, Irsula OR, Lasken RS et al. (2004). Two methods of whole-genome amplification enable accurate genotyping across a 2320-SNP linkage panel. Genome Res 14: 901–907.
Bergen AW, Qi Y, Haque KA, Welch RA, Chanock SJ . (2005). Effects of DNA mass on multiple displacement whole genome amplification and genotyping performance. BMC Biotechnol 5: 24.
Blanco L, Bernad A, Lazaro JM, Martin G, Garmendia C, Salas M . (1989). Highly efficient DNA synthesis by the phage Φ29 DNA polymerase. Symmetrical mode of DNA replication. J Biol Chem 264: 8935–8940.
Blanco L, Salas M . (1984). Characterization and purification of a phage Φ29-encoded DNA polymerase required for the initiation of replication. Proc Natl Acad Sci USA 81: 5325–5329.
Blanco L, Salas M . (1985). Characterization of a 3′–5′ exonuclease activity in the phage Φ29-encoded DNA polymerase. Nucl Acids Res 13: 1239.
Breitbart M, Rohwer F . (2005). Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques 39: 729–736.
Broderick NA, Raffa KF, Goodman RM, Handelsman J . (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl Environ Microbiol 70: 293–300.
Brodie EL, DeSantis TZ, Parker JPM, Zubietta IX, Piceno YM, Andersen GL . (2007). Urban aerosols harbor diverse and dynamic bacterial populations. Proc Natl Acad Sci USA 104: 299–304.
Cheung VG, Nelson SF . (1996). Whole genome amplification using a degenerate oligonucleotide primer allows hundreds of genotypes to be performed on less than one nanogram of genomic DNA. Proc Natl Acad Sci USA 93: 14676–14679.
Christner BC, Mosley-Thompson E, Thompson LG, Reeve JN . (2001). Isolation of bacteria and 16S rDNAs from Lake Vostok accretion ice. Environ Microbiol 3: 570–577.
Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P et al. (2002). Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci USA 99: 5261–5266.
Dean FB, Nelson JR, Giesler TL, Lasken RS . (2001). Rapid amplification of plasmid and phage DNA using phi29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res 11: 1095–1099.
Detter JC, Jett JM, Lucas SM, Dalin E, Arellano AR, Wang M et al. (2002). Isothermal strand-displacement amplification applications for high-throughput genomics. Genomics 80: 691–698.
Dickson PA, Montgomery GW, Henders A, Campbell MJ, Martin NG, RJames M . (2005). Evaluation of multiple displacement amplification in a 5 cM STR genome-wide scan. Nucl Acids Res 33: e119.
Dunning AM, Talmud P, Humphries SE . (1988). Errors in the polymerase chain reaction. Nucleic Acids Res 16: 10393.
Edwards RA, Rodriguez-Brito B, Wegley L, Haynes M, Breitbart M, Peterson DM et al. (2006). Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 7: 57.
Esteban JA, Salas M, Blanco L . (1993). Fidelity of Φ29 DNA polymerase: comparison between protein-primed initiation and DNA polymerization. J Biol Chem 268: 2719–2726.
Fernández-Ortuño D, Torés J, de Vicente A, Pérez-García A . (2007). Multiple displacement amplification, a powerful tool for molecular genetic analysis of powdery mildew fungi. Curr Genet 51: 209–219.
Foster SJ, Monahan BJ . (2005). Whole genome amplification from filamentous fungi using Phi29-mediated multiple displacement amplification. Fungal Genet Biol 42: 367–375.
Gadkar V, Rillig MC . (2005). Application of Phi29 DNA polymerase mediated whole genome amplification on single spores of arbuscular mycorrhizal (AM) fungi. FEMS Microbiol Lett 342: 65–71.
Gonzalez JM, Portillo MC, Saiz-Jimenez C . (2005). Multiple displacement amplification as a pre-polymerase chain reaction (pre-PCR) to process difficult to amplify samples and low copy number sequences from natural environments. Environ Microbiol 7: 1024–1028.
Gorrochotegui-Escalante N, Black IV WC . (2003). Amplifying whole insect genomes with multiple displacement amplification. Insect Mol Biol 12: 195–200.
Groathouse NA, Brown SE, Knudson DL, Brennan PJ, Slayden RA . (2006). Isothermal amplification and molecular typing of the obligate intracellular pathogen Mycobacterium leprae isolated from tissues of unknown origins. J Clin Microbiol 44: 1502–1508.
Holbrook JF, Stabley D, Sol-Church K . (2005). Exploring whole genome amplification as a DNA recovery tool for molecular genetic studies. J Biomol Tech 16: 125–133.
Hosono S, Faruqi AF, Dean FB, Du Y, Sun Z, Wu X et al. (2003). Unbiased whole-genome amplification directly from clinical samples. Genome Res 13: 954–964.
Huang WE, Stoecker K, Griffiths R, Newbold L, Daims H, Whiteley AS et al. (2007). Raman-FISH: combining stable-isotope Raman spectroscopy and fluorescence in situ hybridization for single cell analysis of identity and function. Environ Microbiol 9: 1878–1889.
Hugenholtz P, Huber T . (2003). Chimeric 16S rDNA sequences of diverse origin are accumulating in the public databases. Int J Syst Evol Microbiol 53: 289–293.
Hutchison III CA, Smith HO, Pfannkoch C, Venter JC . (2005). Cell-free cloning using Φ29 DNA polymerase. Proc Natl Acad Sci USA 102: 17332–17336.
Inoue J, Shigemori Y, Mikawa T . (2006). Improvements of rolling circle amplification (RCA) efficiency and accuracy using Thermus thermophilus SSB mutant protein. Nucleic Acids Res 34: e69.
Ishøy T, Kvist T, Westermann P, Ahring BK . (2006). An improved method for single cell isolation of prokaryotes from meso-, thermo-and hyperthermophilic environments using micromanipulation. Appl Microbiol Biotechnol 69: 510–514.
Jeyaprakash A, Hoy MA . (2004). Multiple displacement amplification in combination with high-fidelity PCR improves detection of bacteria from single females or eggs of Metaseiulus occidentalis (Nesbitt) (Acari: Phytoseiidae). J Invertebr Pathol 86: 111–116.
Jiang Z, Zhang X, Deka R, Jin L . (2005). Genome amplification of single sperm using multiple displacement amplification. Nucleic Acids Res 33: e91.
Kalyuzhnaya MG, Zabinsky R, Bowerman S, Baker DR, Lidstrom ME, Chistoserdova L . (2006). Fluorescence in situ hybridization-flow cytometry-cell sorting-based method for separation and enrichment of type I and type II methanotroph populations. Appl Environ Microbiol 72: 4293–4301.
Kvist T, Ahring BK, Lasken RS, Westermann P . (2007). Specific single-cell isolation and genomic amplification of uncultured microorganisms. Appl Environ Microbiol 74: 926–935.
Lage JM, Leamon JH, Pejovic T, Hamann S, Lacey M, Dillon D et al. (2003). Whole genome analysis of genetic alterations in small DNA samples using hyperbranched strand displacement amplification and array-CGH. Genome Res 13: 294–307.
Lasken RS . (2007). Single-cell genomic sequencing using Multiple Displacement Amplification. Curr Opin Microbiol 10: 1–7.
Lasken RS, Egholm M . (2003). Whole genome amplification: abundant supplies of DNA from precious samples or clinical specimens. Trends Biotechnol 21: 531–535.
Lasken RS, Raghunathan A, Kvist T, Ishøy T, Westermann P, Ahring BK et al. (2005). Multiple displacement amplification of genomic DNA. In: Hughes S, Lasken RS (eds). Whole Genome Amplification: Methods Express. Scion Publishing Ltd: UK. pp 119–147.
Lasken RS, Stockwell TB . (2007). Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol 7: 19.
Leigh MB, Pellizari VH, Uhlik O, Sutka R, Rodrigues J, Ostrom NE et al. (2007). Biphenyl-utilizing bacteria and their functional genes in a pine root zone contaminated with polychlorinated biphenyls (PCBs). ISME J 1: 134–148.
Leininger S, Urich T, Schloter M, Schwark L, Qi J, Nicol GW et al. (2006). Archaea predominate among ammonia-oxidizing prokaryotes in soils. Nature 442: 806–809.
Lovmar L, Fredriksson M, Liljedahl U, Sigurdsson S, Syvanen AC . (2003). Quantitative evaluation by minisequencing and microarrays reveals accurate multiplexed SNP genotyping of whole genome amplified DNA. Nucleic Acids Res 31: e129.
Lu Y, Gioia-Patricola L, Gomez JV, Plummer M, Franceschi S, Kato I et al. (2005). Use of whole genome amplification to rescue DNA from plasma samples. Biotechniques 39: 511–515.
Marcy Y, Ishøy T, Lasken RS, Stockwell TB, Walenz BP, Halpern AL et al. (2007a). Nanoliter reactors improve multiple displacement amplification of genomes from single cells. PLoS Genet 3: e155.
Marcy Y, Ouverney C, Bik EM, Lösekann T, Ivanova N, Martin HG et al. (2007b). Dissecting biological ‘dark matter’ with single-cell genetic analysis of rare and uncultivated TM7 microbes from the human mouth. Proc Natl Acad Sci USA 104: 11889–11894.
Mavingui P, Tran Van V, Labeyrie E, Rances E, Vavre F, Simonet P . (2005). Efficient procedure for purification of obligate intracellular Wolbachia pipientis and representative amplification of its genome by multiple-displacement amplification. Appl Environ Microbiol 71: 6910–6917.
Mußmann M, Hu FZ, Richter M, de Beer D, Preisler A, Jørgensen BB et al. (2007). Insights into the genome of large sulfur bacteria revealed by analysis of single filaments. PLoS Biol 5: e230.
Nelson JR, Cai YC, Giesler TL, Farchaus JW, Sundaram ST, Ortiz-Rivera M et al. (2002). TempliPhi, phi29 DNA polymerase based rolling circle amplification of templates for DNA sequencing. Biotechniques 32: 44–47.
Neufeld JD, Chen Y, Dumont MG, Murrell JC . (2008). Marine methylotrophs revealed by stable-isotope probing, multiple displacement amplification and metagenomics. Environ Microbiol (doi:10.1111/j.1462-2920.2008.01568.x).
Neufeld JD, Wagner M, Murrell JC . (2007). Who eats what, where and when? Isotope-labelling experiments are coming of age. ISME J 1: 103–110.
Ochsenreiter T, Selezi D, Quaiser A, Bonch-Osmolovskaya L, Schleper C . (2003). Diversity and abundance of Crenarchaeota in terrestrial habitats studied by 16S RNA surveys and real time PCR. Environ Microbiol 5: 787–797.
Paez JG, Lin M, Beroukhim R, Lee JC, Zhao X, Richter DJ et al. (2004). Genome coverage and sequence fidelity of Φ29 polymerase-based multiple strand displacement whole genome amplification. Nucl Acids Res 32: e71.
Paunio T, Reima I, Syvanen AC . (1996). Preimplantation diagnosis by whole-genome amplification, PCR amplification, and solid-phase minisequencing of blastomere DNA. Clin Chem 42: 1382–1390.
Podar M, Abulencia CB, Walcher M, Hutchison D, Zengler K, Garcia JA et al. (2007). Targeted access to the genomes of low-abundance organisms in complex microbial communities. Appl Environ Microbiol 73: 3205–3214.
Quaiser A, Ochsenreiter T, Klenk HP, Kletzin A, Treusch AH, Meurer G et al. (2002). First insight into the genome of an uncultivated crenarchaeote from soil. Environ Microbiol 4: 603–611.
Raghunathan A, Ferguson HR, Bornarth CJ, Song W, Driscoll M, Lasken RS . (2004). Genomic DNA amplification from a single bacterium. Appl Environ Microbiol 71: 3342–3347.
Rönn AC, Andrés O, Bruford M, Crouau-Roy B, Doxiadis G, Domingo-Roura X et al. (2006). Multiple displacement amplification for generating an unlimited source of DNA for genotyping in nonhuman primate species. Int J Primatol 27: 1145–1169.
Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S et al. (2007). The Sorcerer II global ocean sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5: e77.
Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT et al. (1988). Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 239: 487–491.
Simon HM, Jahn CE, Bergerud LT, Sliwinski MK, Weimer PJ, Willis DK et al. (2005). Cultivation of mesophilic soil crenarchaeotes in enrichment cultures from plant roots. Appl Environ Microbiol 71: 4751–4760.
Sogin ML, Morrison HG, Huber JA, Welch DM, Huse SM, Neal PR et al. (2006). Microbial diversity in the deep sea and the underexplored ‘rare biosphere’. Proc Natl Acad Sci USA 103: 12115–12120.
Spits C, Le Caignec C, De Rycke M, Van Haute L, Van Steirteghem A, Liebaers I et al. (2006). Optimization and evaluation of single-cell whole-genome multiple displacement amplification. Hum Mutat 27: 496–503.
Stepanauskas R, Sieracki ME . (2007). Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc Nat Acad Sci USA 104: 9052–9057.
Steven B, Léveillé R, Pollard WH, Whyte LG . (2006). Microbial ecology and biodiversity in permafrost. Extremophiles 10: 259–267.
Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A . (1992). Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 13: 718–725.
Teske AP . (2005). The deep subsurface biosphere is alive and well. Trends Microbiol 13: 402–404.
Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW et al. (2005). Comparative metagenomics of microbial communities. Science 308: 554–557.
Vora GJ, Meador CE, Stenger DA, Andreadis JD . (2004). Nucleic acid amplification strategies for DNA microarray-based pathogen detection. Appl Environ Microbiol 70: 3047–3054.
Watabe K, Leusch MS, Ito J . (1984). A 3′–5′ exonuclease activity is associated with phage Φ29 DNA polymerase. Biochem Biophys Res Commun 123: 1019–1026.
Wegley L, Edwards R, Rodriguez-Brito B, Liu H, Rohwer F . (2007). Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ Microbiol 9: 2707–2719.
Wu L, Liu X, Schadt CW, Zhou J . (2006). Microarray-based analysis of subnanogram quantities of microbial community DNAs by using whole-community genome amplification. Appl Environ Microbiol 72: 4931–4941.
Yokouchi H, Fukuoka Y, Mukoyama D, Calugay R, Takeyama H, Matsunaga T . (2006). Whole-metagenome amplification of a microbial community associated with scleractinian coral by multiple displacement amplification using Φ29 polymerase. Environ Microbiol 8: 1155–1163.
Zhang K, Martiny AC, Reppas NB, Barry KW, Malek J, Chisholm SW et al. (2006). Sequencing genomes from single cells by polymerase cloning. Nat Biotechnol 24: 680–686.
Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N . (1992). Whole genome amplification from a single cell: implications for genetic analysis. Proc Natl Acad Sci USA 89: 5847–5851.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Binga, E., Lasken, R. & Neufeld, J. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J 2, 233–241 (2008). https://doi.org/10.1038/ismej.2008.10
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2008.10
Keywords
This article is cited by
-
A remarkably diverse and well-organized virus community in a filter-feeding oyster
Microbiome (2023)
-
Interpersonal variability of the human gut virome confounds disease signal detection in IBD
Communications Biology (2023)
-
Characterization of the human gut virome in metabolic and autoimmune diseases
Inflammation and Regeneration (2022)
-
Metagenomics-enabled microbial surveillance
Nature Microbiology (2022)
-
The gut virome in two indigenous populations from Malaysia
Scientific Reports (2022)
