
Abstract
Addiction to a wide range of substances of abuse has been suggested to reflect a ‘Reward Deficiency Syndrome’. That is, drugs are said to stimulate the reward mechanisms so intensely that, to compensate, the population of dopamine D2 receptors (DD2R) declines. The result is that an increased intake is necessary to experience the same degree of reward. Without an additional intake, cravings and withdrawal symptoms result. A suggestion is that food addiction, in a similar manner to drugs of abuse, decrease DD2R. The role of DD2R in obesity was therefore examined by examining the association between body mass index (BMI) and the Taq1A polymorphism, as the A1 allele is associated with a 30–40% lower number of DD2R, and is a risk factor for drug addiction. If a lower density of DD2R is indicative of physical addiction, it was argued that if food addiction occurs, those with the A1 allele should have a higher BMI. A systematic review found 33 studies that compared the BMI of those who did and did not have the A1 allele. A meta-analysis of the studies compared those with (A1/A1 and A1/A2) or without (A2/A2) the A1 allele; no difference in BMI was found (standardized mean difference 0.004 (s.e. 0.021), variance 0.000, Z=0.196, P<0.845). It was concluded that there was no support for a reward deficiency theory of food addiction. In contrast, there are several reports that those with the A1 allele are less able to benefit from an intervention that aimed to reduce weight, possibly a reflection of increased impulsivity.
Similar content being viewed by others

Introduction
The term food addiction is widely and increasingly used when considering the increase in the incidence of obesity.1 In part at least, this reflects the use of the term in different ways. Analogous to drugs of abuse it has been suggested that certain foods, or certain ingredients, can hijack the functioning of the brain. Alternatively, the term is used in the sense of psychological dependence, perhaps a reflection of a personality that is unable to deal psychologically with the continuous opportunities to eat that are offered by western societies.
Addiction to many drugs of abuse is characterized by a decreased population of dopamine D2 receptors (DD2R) in the striatum, a phenomenon that has usually been interpreted as evidence of decreased dopaminergic activity, although it is not necessarily the case that activity is downregulated. The Taq1A polymorphism (rs1800497) is associated with differences in the number of DDR2 receptors, with those with the A1 allele having a lower density of DDR2 receptors, a risk factor for various physical addictions.2, 3, 4, 5 Hence, if obesity reflects an addiction, there should be a homologous mechanism in those who are overweight. A systematic review is therefore presented of the association between obesity and Taq1A (rs1800497). Although there are several widely quoted reports that the A1 allele is associated with obesity,6, 7 the literature has not, to date, been subject to systematic examination. It was found that a lower population of DD2R does not itself necessarily result in a higher body mass index (BMI). However, in those already obese, it is possible that carrying the A1 allele may be a risk factor for putting on more weight, probably for psychological reasons.
A seminal paper played an important role in developing the suggestion that there is a parallel between obesity and the brain’s response to drugs of abuse.8 There is considerable agreement that addiction to opiates,9 alcohol,10 nicotine,11 cocaine12 and methamphetamine13 are all associated with a decreased number of DD2R in the striatum. Thus, the finding of Wang et al.8 that in a group with an average BMI of 51.2 kg m−2, those with fewer DD2R were more obese, suggested a homology with those addicted to drugs of abuse. This led to the theory that an insensitive reward system generates the need to overeat and in this way increases the release of dopamine.14 A Reward Deficiency Syndrome, a reflection of a low density of DD2R, has been proposed to underlie many types of addiction, including overeating.15 The suggestion is that the consumption of highly palatable food substances stimulates the reward mechanisms of the brain so intensely that, to compensate, the population of DD2R is reduced. The supposed consequence is that the brain now requires a greater degree of stimulation to experience the same degree of reward; that is, additional food needs to be eaten to avoid food cravings and withdrawal symptoms.
This theory that the density of DD2R receptors plays a critical role in addiction can be tested by considering Taq1A. Those with the A1 variant (A1/A1 or A1/A2) have a 30–40% lower density of DD2R.16, 17, 18 In other areas of study a consideration of the A1 allele has supported the suggested role of DD2R receptors in addiction: the A1 allele has been found to be associated with an increased risk of alcoholism,2 opioid dependence,3 responding to cocaine4 and smoking.5 Such data led naturally to the prediction that if obesity reflects physical addiction, then a low level of DD2R receptors, and hence those carrying the A1 allele, should be at risk of putting on weight. A systematic review is presented of the association between the A1 alleles and BMI to establish the role, in obesity, of differences in the population of DD2R.
Materials and methods
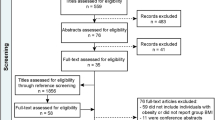
Searches for studies were made both electronically and by following up references quoted in relevant papers. The following databases were searched for studies published in English up to 31 May 2015: PubMed, Web of Knowledge and Google Scholar. The search terms, used as inclusion criteria, were Taq1A, rs1800497, BMI, body weight and obesity. After the initial collection of studies, duplicates were removed and human studies that related Taq1A alleles to BMI, or compared these alleles in groups differing in BMI, were retained. Those of different ages and different ranges of BMI were distinguished to establish whether any effect depended on these parameters. The selection of search terms reflected a precise and focused objective and an interest in a single outcome measure. The progress of the search is outlined in Figure 1. The abstracts of the studies initially identified were examined for duplicates. The abstracts were filtered for those that potentially met the inclusion criteria. The full article of those that remained was then read to establish whether they met the inclusion criteria. Where the reported data allowed it, the BMI of those with the various alleles are listed. When those who were obese were compared with a control group of lesser weight, the frequency of the various alleles in these two groups was reported.

The progressive identification and selection of studies that associated Taq1A to BMI.
Meta-analyses were calculated using the Comprehensive Meta-analysis statistical package (Biostat, Englewood, NJ, USA). A random-effect model rather than fixed-effect model was used in these analyses, as it was not justified to assume that there was only one true effect size. That is, the effect may differ with age or BMI. In addition, those addicted to other substances, or those with morbid obesity, may potentially represent different samples.
Results
In total, 33 studies were found that related Taq1A to BMI. For clarity the studies have been grouped into those dealing with children and adolescents: adults with an acceptable BMI or who were overweight; those who were obese with a BMI between 30 and 40 kg m−2; and those with a BMI >40 kg m−2. Distinguishing studies in this way allowed the influence of the density of dopamine receptors to be examined at different stages of life and in groups of increasing BMI. It has been suggested that DDR2 may play a particular role in those morbidly obese.
Table 1 lists studies of children and adolescents. The pattern was completely consistent. No study reported that the BMI of those with or without the A1 allele differed.19, 20, 21, 22, 23, 24 Similarly, the percentage of those with and without the A1 allele did not differ in groups created because they were or were not obese.20, 25, 26, 27 The study of Hardman et al.23 was particularly instructive as it was a prospective study with a large sample, chosen to be representative of the general population. Pregnant women were recruited and their children followed up for 11 years. The BMI and waist circumference of the resulting children did not differ at ages 7, 8, 9, 10 and 11 years, depending on Taq1A. Similarly, the mothers weight when assessed before pregnancy, and again after 7, 8, 9 and 11 years, was never related to the genetic makeup.
Data associated with adults with a healthy BMI, or who were overweight, are found in Table 2. The vast majority of studies again found that the BMI of those with and without the A1 allele did not differ.19, 22, 23, 24, 28, 29, 30, 31, 32, 33, 34 In contrast to these 11 studies, there was only one report that the A1 allele was associated with a higher BMI.35 A concern with this single positive finding was that the sample considered smokers, a group likely to have a higher incidence of the A1 allele.5 With the case–control approach the findings of the incidence of A1 allele in groups who either were or were not obese were equivocal. Although some found that the incidence of the A1 allele differed in those who had been selected for their heavier weight,28, 29, 36 others did not.32, 37, 38, 39
Data related to those who were obese can be found in Table 3. Again, almost universally those who compared the obese with controls of healthy weight reported that the BMI of those with and without the A1 allele did not differ.7, 40, 41, 42, 43 Davis et al.44 did, however, find a higher incidence of the A1 allele in a sample of the obese as compared with those with binge eating disorder, although both samples were of similar weight (BMI 38.7 and 38.6 kg m−2). There was only one study that found the incidence of A1 to be higher in the obese.7 Various questions arise because, of the 33 studies in this review, it was the only one that reports a group where 100% had the A2/A2 allele, a reflection of extreme inclusion criteria that made the control group unrepresentative of the general population. In addition, a majority of the obese sample had a history of drug abuse that itself was reported to be associated with a higher incidence of the A1 allele.7
Finally, as some have proposed that DD2R may play a particular role in the development of extreme obesity, data associated with those with a BMI >40 kg m−2 were collated (Table 4). When considering the relationship between the various alleles and BMI, the data were equivocal. Although one study reported a higher incidence of the A1 allele in the morbidly obese,6 most did not.32, 38, 39, 45
Figure 2 presents a meta-analysis of the studies, where the BMI was available, of those with (A1/A1 and A1/A2) or without (A2/A2) the A1 allele. There was virtually no difference in BMI depending on the presence of the allele (standardized mean difference 0.004 (s.e. 0.021), variance 0.000, Z=0.196, P<0.845). As one study23 had a sample size larger than the other studies put together, an analysis was calculated with that study removed to ensure that the finding was representative of the entire group of studies. However, the finding was very similar (standardized mean difference 0.004 (s.e. 0.044), variance 0.002, Z=0.102, P<0.919). When those with a BMI >30 kg m−2 were considered,32, 40, 42, 43, 46, 47 again the A1 allele was not associated with BMI (standardized mean difference 0.035 (s.e. 0.085), variance 0.007, lower to higher limit 0.007–0.137, Z=0.405, P<0.686). Similarly, the allele was not associated with BMI if those with a BMI >40 kg m−2 (refs. 32, 46, 47) were selectively examined (standardized mean difference 0.068 (s.e. 0.124), variance 0.015, lower to higher limit −0.175 to 0.310, Z=0.545, P<0.586).

Meta-analysis of the BMI of those with and without the Taq1A allele. The bars report the 95% CI with a central block proportional to the study size. The pooled effect size estimate is reported as a diamond.
Another way of considering the data was to compare the frequency of the A1 allele in a group of obese as compared with a group with a BMI <25 kg m−2. There were 10 studies that had presented such data. Of these 10 studies, 9 were used for the analysis. The study of Blum et al.7 was excluded because the distribution of the alleles of Taq1A was unlike any other study (Table 3): whereas other studies found at least 50% of cases had at least one A1 allele, the study of Blum et al.7 used extreme inclusion criteria and had no incidence of the A1 allele. As such, these data could not be creditably generalized to any normal population. Figure 3 reports a meta-analysis of the frequency that the A1 allele was found in samples of those with a BMI <25 kg m−2 as compared with those who were obese. Overall, there was a greater chance of having the A1 allele if in the obese group (Z=2.005, P<0.045, odds ratio (OR)=1.446, 95% confidence interval (CI) 1.008–2.073). However, when the three studies of children and adolescents were considered alone, there was no significant difference and the overall OR trended toward a higher frequency of the A1 allele in those who were not obese (Z=−0.331, P<0.741, OR=0.890, 95% CI 0.446–1.775). Similarly, a consideration of the 6 studies of adults just failed to demonstrate an overall difference in the incidence of the A1 allele (Z=1.789, P<0.070, OR=1.632, 95% CI 0.954–2.790). An important aspect of the overall data set is that any significance relied greatly on the study of Chen et al.36 that had an OR of 4.1. Although a study should not be removed in this type of analysis, it may be relevant that the other eight resulted in a nonsignificant finding (Z=1.644, P<0.100, OR=1.275, 95% CI 0.954–1.644). Finally, the possibility was considered that the A1 allele was specifically associated with those who were morbidly obese. Three studies were therefore analyzed with an average BMI in the range of 38–40 kg m−2.32, 39, 45 No significant association was found, with the trend being slightly in the direction opposite to that predicted by the reward deficiency hypothesis (Z=−0.170, P<0.865, OR=0.938, 95% CI 0.447–1.966).

Meta-analysis of case–control studies of the incidence of the Taq1A allele in those with a BMI <25 or >30 kg m−2. The bars report the 95% CI with a central block proportional to the study size. The pooled effect size estimate is reported as a diamond.
Discussion
To a large extent the suggestion that food can be addictive has relied on data generated using various brain imaging techniques. Given that many of those carrying out these studies have a background in the study of drugs of abuse, they have naturally used addiction as an explanation of their findings. Such data, however, need to be put into context. It is received wisdom that drugs of abuse act by hijacking the sites that mediate natural rewards, such as food or sex, sites that are mediated by dopaminergic mechanisms. Hence, any demonstration that an interest in food influences dopaminergic activity cannot necessarily be taken as evidence of addiction, rather it needs to be demonstrated that a response is abnormal.
A concern is that although dopamine has a well-described role in reward mechanisms, it also influences other aspects of behavior that could potentially contribute to weight gain. It is incumbent on those proposing addiction as a mechanism to exclude alternative explanations. It is argued below that any lower density of dopamine receptors in the obese is better explained as producing differences in personality, rather than as an indication of physical addiction. The 33 studies that had related Taq1A to BMI produced remarkably consistent findings. In children and adolescents, there was no association between the presence of the A1 allele and BMI (Table 1). In those with a BMI between 30 and 40 kg m−2 (Table 3), it did not differ depending on the A1 allele. Finally, in those with BMI up to 30 kg m−2 (Table 2), there was an association in only 1 of 13 samples. When the BMI was in excess of 40 kg m−2, there were two studies that again found that the A1 allele was without influence,32, 46 although Carpenter et al.47 found a significant difference. In total, of the 29 samples only 2 reported that there was a difference in weight associated with the presence of Taq1A,35, 45, 48 although the relationship in the study of Morton et al.35 may plausibly reflect a group chosen for a history of addiction. The combination of these findings into a meta-analysis (Figure 2) found no overall significant difference in the BMI of those with or without the A1 allele. Although the frequency of the A1 allele was found to be greater when groups with and without obesity were compared (Figure 3), the effect was limited and depended greatly on one study.36 It is likely that in these case–control studies much depended on how the sample of the obese was recruited. If, as discussed below, the A1 allele did not cause obesity, but rather made it difficult to deal with it, the recruitment of a sample of those already obese may have artificially raised the incidence of the A1 allele.
The obvious conclusion was that in the vast majority, if not the entire population, the density of DD2R is not associated with the development of obesity. These findings (Figure 2) suggest a difference between the mechanisms that underlie obesity and addiction to drugs of abuse, as in the latter the A1 allele has been found to be a risk factor for addiction.2, 3, 4, 5 The findings gave no support for a DDR2-related reward deficiency syndrome as a mechanism that underlies obesity.
Yet, tentatively it may be proposed that there are rare instances of the A1 allele predisposing to morbid obesity, although the data are limited and contradictory. Carpenter et al.47 found in those with a BMI of 43 kg m−2 that the A1 allele was associated with a difference in weight. Similarly, Wang et al.8 reported in those with an average BMI of 51 kg m−2 that a lower density of DD2R was associated with greater weight. However, these findings can be viewed as consistent with the view considered below that the A1 allele was not responsible for obesity, but rather predisposes to a personality that finds it difficult to respond to attempts to reduce weight. However, the manner in which the population of the morbidly obese is selected may be critical, as it has also been found that the morbidly obese did not differ in the incidence of the A1 allele32, 46 or the density of DD2R.48, 49, 50, 51
As the present summary is at variance with the suggestion that there is a lower density of DD2R in the obese, the seminal work of Wang et al.8 is considered, as it is quoted frequently when the possibility of food addiction is proposed. Using positron emission tomography, they used radiolabeled raclopride, a dopamine antagonist, to measure the density of striatal DD2R and found it was lower in a group of 10 obese individuals. In the obese, but not the control group, the BMI correlated negatively with the number of DD2R. The findings were interpreted as evidence that ‘dopamine deficiency in obese individuals may perpetuate pathological eating as a means to compensate for decreased activation of these circuits’. However, the obese group had an average BMI of 51 kg m−2 as compared with a control group with an average BMI of 25 kg m−2. Although these findings have been used to support the view that diet-induced changes in dopaminergic mechanisms have a role in the obesity epidemic, the findings from such an extreme group should not be generalized uncritically to the general population. In addition, although in this extreme sample the density of DD2R was less in the more obese, a majority of those in the nonobese sample had a level of DD2R similar to those with a BMI of >50 kg m−2. It was clear that differences in dopaminergic mechanisms did not inevitably lead to being overweight. If a low density of D2 receptors is the mechanism behind ‘pathological eating’, why was it not influential in everybody?
How should the findings of Wang et al.8 be interpreted? It was apparent that a low density of DD2R is compatible with being both of a healthy weight or morbidly obese; a finding supported by the studies that have considered Taq1A (Tables 1–4). Yet, in the obese group a low density of DD2R was associated with a greater body mass,8 an observation that suggested that some additional DD2R-related mechanism influenced those who are already overweight. These findings8 should, however, be viewed with considerable caution as they have not been universally replicated. Whereas some studies52, 53, 54 have supported the findings of Wang et al.8 others have not.48, 49, 50, 51 In fact, there are even reports that DD2R is higher in those who are obese,9, 55 and that in some areas of the brain, as BMI increased the levels of DD2R increased rather than decreased.56
As dopamine modulates a range of behaviors other than those associated with physical addiction, it is plausible that the influence of the density of DDR2 is mediated via other mechanisms. Figure 4 outlines two dopaminergic pathways that both originate in the ventral tegmental area. The mesolimbic tract is particularly involved in the experience of reward and pleasure and is known to be a site important in the action of drugs of abuse. In contrast, the mesocortical tract innervates the frontal cortex, an area involved in executive functioning, motivation and the planning of behavior. There is growing evidence that behavior associated with differences in density of DD2R reflects the activity of the mesocortical, rather than mesolimbic, pathway. In morbidly obese but not control subjects, lower levels of striatal DD2R were positively correlated with metabolism in various areas of the frontal lobes.54 It was concluded that ‘decreases in striatal D2 receptors could contribute to overeating via their modulation of striatal prefrontal pathways, which participate in inhibitory control and salience attribution’.

The mesolimbic and mesocortical dopaminergic pathways. Two pathways that are mediated by dopamine are illustrated. The mesolimbic pathway is particularly associated with reward and pleasure and is associated with addiction to cocaine, alcohol and nicotine. The mesocortical pathway leads to the frontal cortex and is involved with planning and executive functioning.
Nisoli et al.32 concluded that ‘the presence of A1 allele is not simply related to body weight but the A1 allele might be a marker of a genetic psychological condition in people with high risk to develop pathological eating behaviour’. They found the A1 allele was associated with a preoccupation with gaining weight that was combined with a feeling of not having control over your life. The A1 allele has also been associated with novelty or sensation seeking,57 performance of a delayed discounting task 58 and impulsivity on a card sorting task.59, 60 Those carrying A1 have been reported to be less able to learn to avoid behavior with negative consequences.61, 62 White et al.59 concluded that the A1 allele was associated with a ‘rash impulsive behavioural style and reinforcement-related learning deficits’. Ariza et al.45 similarly found that carrying the A1 allele, but only when obese, ‘could confer a weakness as regards the performance of executive functions’. It is easy to see that if the A1 allele was associated with impulsivity, risk taking and seeking reward, when combined with a decreased ability to learn from any negative consequences of such behavior, this would result in an inability to respond appropriately to any attempt to reduce food intake.
Consistent with the view that in the obese, the A1 allele is associated with a psychological profile that makes it difficult to deal with multiple opportunities to eat, there is an increasing number of reports that the A1 allele is associated with an inability to benefit from attempts to lose weight. Roth et al.22 reported that Taq1A influenced the response of obese children who were taking part in a 1-year intervention: those with A1/A1 gained weight over the year, whereas those with other genotypes lost weight. Similarly, obese post-menopausal women with the A1 allele lost less weight when on a diet than those with the A2 variant.43 Again, Winkler et al.46 reported that those with the A1 allele were less able to maintain weight loss after dieting. In a sample of white but not black diabetics, Barnard et al.42 found that those with the A1 allele were less able to reduce their fat intake when put on a low-fat diet. In fact, in all four of these studies, the Taq1A gene was not associated with baseline body weight. In the longer term, a life time of living with the A1 allele was not associated with a greater BMI, although in the short term it affects attempts to lose weight.
In addition, there is evidence that the A1 allele increases the response to food. Stice et al.,21 in a brain scanner, monitored the response to pictures of foods that were or were not appetizing. In those with the A1 allele, a smaller response to palatable foods was associated with a greater rise in weight over the subsequent year. It was suggested on this basis that ‘individuals may overeat to compensate for a hypo-functioning dorsal striatum, particularly in those with genetic polymorphisms thought to attenuate dopamine signalling in this region’. However, although the baseline BMI was not reported, it proved possible to calculate these data from the information provided. Consistent with the present analysis, an increase in weight in those with the A1 allele was only associated with the experimental period: at baseline, the BMI did not differ (A1 allele: BMI=23.3 kg m−2; no A1 allele: BMI=24.8 kg m−2). Over the previous 15.6 years, differences in the density of DDR2 had made no difference to body weight. Rather than supporting the view that a low density of DD2R encourages overeating, the findings of Stice et al.,21 were compatible with the present conclusion that the polymorphisms of Taq1A (Tables 1–3) do not in the long term influence the BMI of the general population.
Thus, the area is characterized by two apparently contradictory findings. The evidence is overwhelming that in the general population the A1 allele is not associated with differences in BMI (Tables 1–4). Yet, there are reports that in experimental situations the A1 allele is associated with a lesser ability to benefit from an intervention that aimed to decrease weight.22, 42, 43, 46 The question then arises as how a lower density of DD2R can fail to be associated with higher baseline levels of BMI when it makes losing weight more difficult.
An explanation is that isolated findings from brain imaging studies should not be uncritically generalized to real-world conditions. Any theory of the cause of obesity based on a single limited type of data is almost certain to be inadequate. When the UK government asked for the multitude of factors involved in obesity to be listed, 110 general factors were found, each of considerable complexity, with so many interactions that the diagrams produced were reminiscent of spaghetti.62 The factors fell into 10 general categories: biological, media, social, psychological, economic, food, activity, infrastructure, developmental and medical. As such, it is inherently improbable, if not impossible, that any meaningful understanding of obesity is going to result from a single isolated conception of the problem. Although those using imaging techniques tend to acknowledge the complexity of the situation, the development of a conception such as food addiction risks offering a very simple explanation of an extremely complex problem.
Any theory arising from brain imaging should not be tested in yet more brain imaging studies: rather, to gain credibility it needs to be tested using other approaches and related to the body weight of those living under real-world conditions. As an example, Benton63 developed a series of predictions that would be true if sugar addiction occurred; for example, that tolerance would develop and withdrawal symptoms would be generated by opiate antagonists. When over a dozen predicted consequences of food addiction were examined, on no occasion was a prediction supported. Similarly, in the present review, the theory that a lower density of DD2R is associated with an increase in BMI gained little support from examining those not taking part in an experiment (Tables 1–4). In contrast, when subject to a dietary intervention that aims to decrease body weight, many times a day an individual has to consciously make decisions about what to eat. Food choice to a large extent reflects automated unconscious decisions: every day we eat the same breakfast or the same sandwich for lunch. However, when on a diet, or taking part in a clinical trial, many times a day the choice of food becomes both conscious and salient. In these circumstances the influence of preexisting behavioral predispositions becomes critical.
The present conclusions have similarities with those of Neurofast,64 a European Union-funded consortium of workers in eight countries charged with evaluating the evidence behind suggested food addiction. After reviewing the topic from a wide range of perspectives, they concluded that food addiction was unlikely to underlie most instances of obesity, as it usually reflects a long-term marginal overconsumption of calories. Rather, the conclusion was that ‘eating addiction’ was a better conception. The implication was that a psychological compulsion to eat might develop: that is, obesity is better seen as a behavioral disorder. However, the present conclusions about DD2R do not preclude the obvious fact that some individuals have a serious problem in controlling food intake. The important question is the underlying mechanism and hence the best manner by which obesity can be addressed.
A suggested role for ‘eating addiction’, rather than ‘food addiction’,64 implies that obesity should not be addressed by concentrating on food itself, but rather the individual’s relationship with eating. ‘Eating addiction’ stresses the behavioral component, whereas ‘food addiction’ is a passive process that simply befalls the individual, a consequence of the ready availability of palatable foods. If differences in the ability to deal with food-related issues can be demonstrated, a possibility that should be considered is that obesity might be better addressed by offering different intervention strategies to those with different genetic makeups.
What implications do the present findings have when developing a response to obesity? Although imaging studies that have looked at the role of dopaminergic mechanisms report a greater response to palatable foods,19, 21 the essence of the problem is that realistically individuals are going to choose to eat foods that taste good. We choose to eat palatable foods that stimulate reward mechanisms and in turn more food is consumed. Any approach of removing the constituents that make food palatable is extremely unlikely to work. For example, the wide-spread decision to produce low-fat products did not prevent the progressive increase in the incidence of obesity. The consumer can choose what they eat and will mostly choose tasty food: no food manufacturer or restaurant chain is going to survive by providing foods that are not palatable. Typically, palatability reflects the content of fat and sugar, often in combination. However, a recent review confirmed ‘the existence of the sugar-fat seesaw on a percentage energy basis’; 65 that is, diets high in fat tend to be low in sugar and vice versa. Part of this phenomenon may reflect the desire for palatability, so that the removal of one palatable dietary constituent leads to the consumption of another. This finding suggests that attempts to address the incidence of obesity by decreasing palatability are unlikely to succeed. If one wishes to decrease energy density while maintaining palatability, particular attention should be given to fat because, of the various macronutrients, it offers the most energy per gram and is the least able to induce satiety.66
Rather than taking the ‘addiction’ approach of decreasing particular nutrients, the aim should be to offer foods that taste good, that are satiating and yet provide less energy. The various macronutrients generate different levels of satiety;66 low-energy-dense diets generate greater satiety; high-energy-dense foods lead to ‘passive overconsumption’, that is, as the food lacks bulk, excess energy is unintentionally consumed.67 The World Health Organisation68 concluded that a key cause of obesity was the increased consumption of energy-dense foods: high-energy-dense foods are those that contain the least water and the most fat.67
Such a conclusion does not get the food industry off the hook but rather sets a different agenda. A ‘food addiction’ approach would involve establishing the addictive substance and reducing the amount in food, be it fat, sugar a combination of these or some other ingredients. However, when taking the ‘eating addiction’ perspective you cannot escape the obvious fact that obesity largely reflects the extensive availability of highly palatable and highly calorific foods. The task for the food industry is to aid energy restriction by producing low-energy-dense foods that are formulated to be palatable (or nobody will buy them), maximize satiety and prolong satiation. Even so, such is the multitude of factors that influence obesity that a dietary approach is unlikely to have a meaningful impact unless it is part of a coordinated program that acknowledges the complexity and multifaceted nature of the problem. Although an increased provision of low-energy-dense foods would be helpful, the message is that a change in the nature of food items will not by itself be a sufficient response. The present analysis suggests that one aspect of such an integrated response to obesity should be how individuals deal psychologically with the continual temptations to eat. Specifically, we should consider further whether the strategies that are recommended need to be suited to our genetic makeup and hence our personalities.
References
Brownell KD, Gold MS (eds). Food and Addiction. Oxford University Press: Oxford, UK, 2012.
Munafò MR, Matheson IJ, Flint J . Association of the DRD2 gene Taq1A polymorphism and alcoholism: a meta-analysis of case–control studies and evidence of publication bias. Mol Psychiatry 2007; 12: 454–461.
Deng XD, Jiang H, Ma Y, Gao Q, Zhang B, Mu B et al. Association between DRD2/ANKK1 TaqIA polymorphism and common illicit drug dependence: evidence from a meta-analysis. Hum Immunol 2015; 76: 42–51.
Verdejo-Garcia A, Clark L, Verdejo-Román J, Albein-Urios N, Martinez-Gonzalez JM, Gutierrez B et al. Neural substrates of cognitive flexibility in cocaine and gambling addictions. Br J Psychiatry 2015; 207: 158–164.
Munafo MR, Timpson NJ, David SP, Ebrahim S, Lawlor DA . Association of the DD2R gene Taq1A polymorphism and smoking behavior: a meta-analysis and new data. Nicotine Tob Res 2009; 11: 64–76.
Comings DE, Flanagan SD, Dietz G, Muhleman D, Knell E, Gysin R . The dopamine D2 receptor as a major gene in obesity and height. Biochem Med Metab Biol 1993; 50: 176–185.
Blum K, Braverman ER, Wood RC, Gill J, Li C, Chen TJ et al. Increased prevalence of the Taq I A1 allele of the dopamine receptor gene (DRD2) in obesity with comorbid substance use disorder: a preliminary report. Pharmacogenetics 1996; 6: 297–305.
Wang GJ, Volkow ND, Logan J, Pappas NR, Wong CT, Zhu W et al. Brain dopamine and obesity. Lancet 2001; 357: 354–357.
Martinez D, Saccone PA, Liu F, Slifstein M, Olowska D, Grassetti A et al. Deficits in dopamine D2 receptors and pre-synaptic dopamine in heroin dependence: commonalities and differences with other types of addiction. Biol Psychiatry 2012; 71: 192–198.
Martinez D, Gil R, Slifstein M, Hwang D-R, Huang Y, Perez A et al. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychaitry 2005; 58: 779–786.
Fehr C, Yakushev I, Hohmann N, Buchholz HG, Landvogt C, Deckers H et al. Association of low striatal dopamine D2 receptor availability with nicotine dependence similar to that seen with other drugs of abuse. Am J Psychiatry 2008; 165: 507–514.
Martinez D, Broft A, Foltin RW, Slifstein M, Hwang DR, Huang Y et al. Cocaine dependence and D2 receptor availability in the functional subdivisions of the striatum: relationship with cocaine-seeking behavior. Neuropsychopharmacology 2004; 29: 1190–1202.
Volkow ND, Chang L, Wang GJ, Fowler JS, Ding YS, Sedler M et al. Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry 2001; 158: 2015–2021.
Wang GJ, Volkow ND, Thanos PK, Fowler JS . Similarity between obesity and drug addiction as assessed by neurofunctional imaging: a concept review. J Addict Dis 2004; 23: 39–53.
Blum K, Febo M, McLaughlin T, Cronje FJ, Han D, Gold SM . Hatching the behavioral addiction egg: Reward Deficiency Solution System (RDSS)(TM) as a function of dopaminergic neurogenetics and brain functional connectivity linking all addictions under a common rubric. J Behav Addict 2014; 3: 149–156.
Noble EP, Blum K, Ritchie T, Montgomery A, Sheridan PJ . Allelic association of the D2 dopamine receptor gene with receptor binding characteristics in alcoholism. Arch Gen Psychiatry 1991; 48: 648–654.
Pohjalainen T, Rinne JO, Nagren K, Lehikoinen P, Anttila K, Syvalahti EK et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry 1998; 3: 256–260.
Jonsson EG, Nothen NM, Grunhage F, Farde L, Nakashima Y, Propping P et al. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry 1999; 4: 290–296.
Stice E, Spoor S, Bohon C, Small DM . Relation between obesity and blunted striatal response to food is moderated by TaqIA A1 allele. Science 2008; 322: 449–452.
Ergun MA, Karaoguz MY, Koc A, Camurdan O, Bideci A, Yazici AC et al. The apolipoprotein E gene and Taq1A polymorphisms in childhood obesity. Genet Test Mol Biomarkers 2010; 14: 343–345.
Stice E, Yokum S, Bohon C, Marti N, Smolen A . Reward circuitry responsivity to food predicts future increases in body mass: moderating effects of DD2R and DRD4. Neuroimage 2010; 50: 1618–1625.
Roth CL, Hinney A, Schur EA, Elfers CT, Reinehr T . Association analyses for dopamine receptor gene polymorphisms and weight status in a longitudinal analysis in obese children before and after lifestyle intervention. BMC Pediatr 2013; 13: 197.
Hardman CA, Rogers PJ, Timpson NJ, Munafò MR . Lack of association between DRD2 and OPRM1 genotypes and adiposity. Int J Obes 2014; 38: 730–736.
Yokum S, Marti CN, Smolen A, Stice E . Relation of the multilocus genetic composite reflecting high dopamine signaling capacity to future increases in BMI. Appetite 2015; 87: 38–45.
Duran-Gonzalez J, Ortiz I, Gonzales E, Ruiz N, Ortiz M, Gonzalez A et al. Association study of candidate gene polymorphisms and obesity in a young Mexican-American population from South Texas. Arch Med Res 2011; 42: 523–531.
Araz NC, Nacak M, Balci SO, Benlier N, Araz M, Pehlivan S . Childhood obesity and the role of dopamine D2 receptor and cannabinoid receptor-1 gene polymorphisms. Gen Test Mol Biomarkers 2012; 16: 1408–1412.
Aksyonova E, Seyitnazarova A, Solntsava A, Zagrebaeva O, Mikhno H, Sukalo A . Association analysis of the D2 dopamine receptor gene Taq IA polymorphisms in a cohort of Belarus morbidly obese children and adolescents. Endo Abst 2014; 35: 778.
Spitz MR, Detry MA, Pillow P, Hu YH, Amos CI, Hong WK et al. Variant alleles of the D2 dopamine receptor gene and obesity. Nutr Res 2000; 20: 371–380.
Thomas GN, Critchley JAJH, Tomlinson B, Cockram CS, Chan JC . Relationships between the TaqI polymorphism of the dopamine D2 receptor and blood pressure in hyperglycaemic and normoglycaemic Chinese subjects. Clin Endocrinol 2001; 55: 605–611.
Epstein LH, Wright SM, Paluch RA, Leddy JJ, Hawk LW Jr, Jaroni JL et al. Relation between food reinforcement and dopamine genotypes and its effect on food intake in smokers. Am J Clin Nutr 2004; 80: 82–88.
Fang YJ, Thomas GN, Xu ZL, Fang JQ, Critchley JA, Tomlinson B . An affected pedigree member analysis of linkage between the dopamine D2 receptor gene TaqI polymorphism and obesity and hypertension. Int J Cardiol 2005; 102: 111–116.
Nisoli E, Brunani A, Borgomainerio E, Tonello C, Dioni L, Briscini L et al. D2 dopamine receptor (DRD2) gene Taq1A polymorphism and the eating-related psychological traits in eating disorders (anorexia nervosa and bulimia) and obesity. Eat Weight Disord 2007; 12: 91–96.
Epstein LH, Temple JL, Neaderhiser BJ, Salis R, Erbe RW, Leddy JJ . Food reinforcement, the dopamine D-2 receptor genotype, and energy intake in obese and nonobese humans. Behav Neurosci 2007; 121: 877–886.
Athanasoulia AP, Sievers C, Uhr M, Ising M, Stalla GK, Schneider HJ . The effect of the ANKK1/DRD2 Taq1A polymorphism on weight changes of dopaminergic treatment in prolactinomas. Pituitary 2014; 17: 240–245.
Morton LM, Wang SS, Bergen AW, Chatterjee N, Kvale P, Welch R et al. DRD2 genetic variation in relation to smoking and obesity in the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial. Pharmacogenet Genomics 2006; 16: 901–910.
Chen TJH, Blum K, Kaata G, Braverman E, Pullin D, Downs BV et al. Reviewing the role of putative candidate genes in ‘‘Neurobesigenics’’, a clinical subtype of Reward Deficiency Syndrome (RDS). Gene Ther Mol Biol 2007; 11A: 61–74.
Thomas GN, Tomlinson B, Critchley JA . Modulation of blood pressure and obesity with the dopamine D2 receptor gene TaqI polymorphism. Hypertension 2000; 36: 177–182.
Southon A, Walder K, Sanigorski AM, Zimmet P, Nicholson GC, Kotowicz MA et al. Taq IA and Ser311Cys polymorphisms in the dopamine D2 receptor gene and obesity. Diabet Nutr Metab 2003; 16: 72–76.
Davis C, Levitan RD, Kaplan AS, Carter J, Reid C, Curtis C et al. Reward sensitivity and the D2 dopamine receptor gene: A case-control study of binge eating disorder. Prog Neuro Psychopharmacol Biol Psychiatry 2008; 32: 620–628.
Noble EP, Noble RE, Ritchie T, Syndulko K, Bohlman MC, Noble LA et al. Allelic association of the human D2 dopamine receptor gene with obesity. Int J Eating Disord 1994; 15: 205–217.
Jenkinson CP, Hanson R, Cray K, Wiedrich C, Knowler WC, Bogardus C et al. Association of dopamine D2 receptor polymorphisms Ser311Cys and TaqIA with obesity or type 2 diabetes mellitus in Pima Indians. Int J Obes 2000; 24: 1233–1238.
Barnard ND, Noble EP, Ritchie T, Cohen J, Jenkins DJ, Turner-McGrievy G et al. D2 dopamine receptor Taq1A polymorphism, body weight, and dietary intake in type 2 diabetes. Nutrition 2009; 25: 58–65.
Cameron JD, Riou ME, Tesson F, Goldfield GS, Rabasa-Lhoret R, Brochu M et al. The TaqIA RFLP is associated with attenuated intervention-induced body weight loss and increased carbohydrate intake in post-menopausal obese women. Appetite 2013; 60: 111–116.
Davis C, Levitan RD, Yilmaz Z, Kaplan AS, Carter JC, Kennedy JL . Binge eating disorder and the dopamine D2 receptor: genotypes and sub-phenotypes. Prog Neuropsychopharmacol Biol Psychiatry 2012; 38: 328–335.
Ariza M, Garolera M, Jurado MA, Garcia-Garcia I, Hernan I, Sánchez-Garre C et al. Dopamine genes (DRD2/ANKK1-TaqA1 and DRD4-7R) and executive function: their interaction with obesity. PLoS One 2012; 7: e41482.
Winkler JK, Woehning A, Schultz J-H, Brune M, Beaton N, Challa TD et al. TaqIA polymorphism in dopamine D2 receptor gene complicates weight maintenance in younger obese patients. Nutrition 2012; 28: 996–1001.
Carpenter Cl, Wong AM, Li Z, Noble EP, Heber D . Association of dopamine D2 receptor and leptin receptor genes with clinically severe obesity. Obesity 2013; 21: E467–E473.
Steele KE, Prokopowicz GP, Schweitzer MA, Magunsuon TH, Lidor AO, Kuwabawa H et al. Alterations of central dopamine receptors before and after gastric bypass surgery. Obes Surg 2010; 20: 369–374.
Eisenstein SA, Antenor‐Dorsey JAV, Gredysa DM, Koller JM, Bihun EC, Ranck SA et al. A comparison of D2 receptor specific binding in obese and normal‐weight individuals using PET with (N‐[11C] methyl) benperidol. Synapse 2013; 67: 748–756.
Kessler RM, Zald DH, Ansari MS, Li R, Cowan RL . Changes in dopamine release and dopamine D2/3 receptor levels with the development of mild obesity. Synapse 2014; 68: 317–320.
Karlsson HK, Tuominen L, Tuulari JJ, Hirvonen J, Parkkola R, Helin S et al. Obesity is associated with decreased μ-opioid but unaltered dopamine D2 receptor availability in the brain. J Neurosci 2015; 35: 3959–3965.
de Weijer BA, van de Giessen E, van Amelsvoort TA, Boot E, Braak B, Janssen IM et al. Lower striatal dopamine D2/3 receptor availability in obese compared with non-obese subjects. Eur J Nuc Med Mol Imaging Res 2011; 1: 37.
Haltia LT, Rinne JO, Helin S, Parkkola R, Någren K, Kaasinen V . Effects of intravenous glucose on dopaminergic function in the human brain in vivo. Synapse 2007; 61: 748–756.
Volkow ND, Wang GJ, Telang F, Fowler JS, Thanos PK, Logan J et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: possible contributing factors. Neuroimage 2008; 42: 1537–1543.
Dunn JP, Kessler RM, Feurer ID, Volkow ND, Patterson BW, Ansari MS et al. Relationship of dopamine type 2 receptor binding potential with fasting neuroendocrine hormones and insulin sensitivity in human obesity. Diabet Care 2012; 35: 1105–1111.
Guo J, Simmons WK, Herscovitch P, Martin A, Hall KD . Striatal dopamine D2-like receptor correlation patterns with human obesity and opportunistic eating behavior. Mol Psychiatry 2014; 19: 1078–1084.
Ratsma JE, Stelt O, Schoffelmeer AN, Westerveld A, Boudewijn Gunning W . P3 event-related potential, dopamine D2 receptor A1 allele, and sensation-seeking in adult children of alcoholics. Alcohol Clin Exp Res 2001; 25: 960–967.
Eisenberg DT, MacKillop J, Modi M, Beauchemin J, Dang D, Lisman SA et al. Examining impulsivity as an endophenotype using a behavioral approach: a DRD2 TaqI A and DRD4 48-bp VNTR association study. Behav Brain Functions 2007; 3: 2.
White MJ, Morris CP, Lawford BR, Young RM . Behavioral phenotypes of impulsivity related to the ANKK1 gene are independent of an acute stressor. Behav Brain Funct 2008; 24: 54–63.
Jocham G, Klein TA, Neumann J, von Cramon DY, Reuter M, Ullsperger M . Dopamine DRD2 polymorphism alters reversal learning and associated neural activity. J Neurosci 2009; 29: 3695–3704.
Klein TA, Neumann J, Reuter M, Hennig J, von Cramon DY, Ullsperger M . Genetically determined differences in learning from errors. Science 2007; 318: 1642–1645.
Vandenbroeck P, Goossens J, Clemens M . Tackling Obesities: Futures Choices—Obesity System Atlas. Government Office for Science. Her Majesties Stationary Office: London, 2007.
Benton D . The plausibility of sugar addiction and its role in obesity and eating disorders. Clin Nutr 2010; 29: 288–303.
Hebebrand J, Albayrak O, Adan R, Antel J, Dieguez C, de Jong J et al. “Eating addiction”, rather than “food addiction”, better captures addictive-like eating behaviour. Neurosci Biobehav Rev 2014; 47: 295–306.
Sadler MJ, McNulty H, Gibson S . Sugar-fat seesaw: a systematic review of the evidence. Crit Rev Food Sci Nutr 2015; 55: 338–356.
Holt SH, Miller JC, Petocz P, Farmakalidis E . A satiety index of common foods. Eur J Clin Nutr 1995; 49: 675–690.
Drewnowski A . Energy density, palatability, and satiety: implications for weight control. Nutr Rev 1998; 56: 347–353.
World Health Organization (WHO) Diet, nutrition and the prevention of excess weight gain and obesity. Report of a Joint WHO/FAO Expert Consultation. WHO Technical Report Series No. 916. WHO: Geneva, 2003.
Epstein LH, Dearing KK, Erbe RW . Parent-child concordance of Taq1 A1 allele predicts similarity of parent-child weight loss in behavioral family-based treatment programs. Appetite 2010; 55: 363–366.
Davis CA, Levitan RD, Reid C, Carter JC, Kaplan AS, King N et al. Dopamine for ‘wanting’ and opioids for ‘liking’: a comparison of obese adults with and without binge eating. Obesity 2009; 17: 1220–1225.
Acknowledgements
This article is based on a symposium entitled ‘Sweeteners and Health: Findings from Recent Research and their Impact on Obesity and Related Metabolic Conditions’ presented at the 22nd European Congress on Obesity, Prague, on 7 May 2015 with sponsorship from Rippe Lifestyle Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The article is based on a presentation made at the 22nd European Congress on Obesity, Prague 2015, associated with which the Rippe Lifestyle Institute paid travel expenses and an honorarium. In 2008, DB was commissioned by World Sugar Research Organization to review the literature on putative sugar addiction. DB received grant support from Pepsico for a project on hydration, and HY served as co-investigator. HY declares no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Benton, D., Young, H. A meta-analysis of the relationship between brain dopamine receptors and obesity: a matter of changes in behavior rather than food addiction?. Int J Obes 40 (Suppl 1), S12–S21 (2016). https://doi.org/10.1038/ijo.2016.9
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2016.9
This article is cited by
-
Analysis of Factors Associated with Outcomes of Bariatric Surgery: rs1800497 ANKK1, rs1799732 DRD2 Genetic Polymorphisms, Eating Behavior, Hedonic Hunger, and Depressive Symptoms
Journal of Gastrointestinal Surgery (2023)
-
Being underweight, academic performance and cognitive control in undergraduate women
Archives of Women's Mental Health (2023)
-
Neuropeptides Modulate Feeding via the Dopamine Reward Pathway
Neurochemical Research (2023)
-
High Fat Diet Increases [3H] Flunitrazepam Binding in the Mouse Brain that is Dependent on the Expression of the Dopamine D2 Gene
Neurochemical Research (2022)
-
Associations of the ANKK1 and DRD2 gene polymorphisms with overweight, obesity and hedonic hunger among women from the Northwest of Iran
Eating and Weight Disorders - Studies on Anorexia, Bulimia and Obesity (2021)
