
Abstract
Background:
Obesity is strongly associated with prevalence of obstructive sleep apnea (OSA), and weight loss has been shown to reduce disease severity.
Objective:
To investigate whether liraglutide 3.0 mg reduces OSA severity compared with placebo using the primary end point of change in apnea–hypopnea index (AHI) after 32 weeks. Liraglutide’s weight loss efficacy was also examined.
Subjects/Methods:
In this randomized, double-blind trial, non-diabetic participants with obesity who had moderate (AHI 15–29.9 events h−1) or severe (AHI ⩾30 events h−1) OSA and were unwilling/unable to use continuous positive airway pressure therapy were randomized for 32 weeks to liraglutide 3.0 mg (n=180) or placebo (n=179), both as adjunct to diet (500 kcal day−1 deficit) and exercise. Baseline characteristics were similar between groups (mean age 48.5 years, males 71.9%, AHI 49.2 events h−1, severe OSA 67.1%, body weight 117.6 kg, body mass index 39.1 kg m−2, prediabetes 63.2%, HbA1c 5.7%).
Results:
After 32 weeks, the mean reduction in AHI was greater with liraglutide than with placebo (−12.2 vs −6.1 events h−1, estimated treatment difference: −6.1 events h−1 (95% confidence interval (CI), −11.0 to −1.2), P=0.0150). Liraglutide produced greater mean percentage weight loss compared with placebo (−5.7% vs −1.6%, estimated treatment difference: −4.2% (95% CI, −5.2 to −3.1%), P<0.0001). A statistically significant association between the degree of weight loss and improvement in OSA end points (P<0.01, all) was demonstrated post hoc. Greater reductions in glycated hemoglobin (HbA1c) and systolic blood pressure (SBP) were seen with liraglutide versus placebo (both P<0.001). The safety profile of liraglutide 3.0 mg was similar to that seen with doses ⩽1.8 mg.
Conclusions:
As an adjunct to diet and exercise, liraglutide 3.0 mg was generally well tolerated and produced significantly greater reductions than placebo in AHI, body weight, SBP and HbA1c in participants with obesity and moderate/severe OSA. The results confirm that weight loss improves OSA-related parameters.
Similar content being viewed by others


Introduction
Obstructive sleep apnea (OSA) is a prevalent yet underdiagnosed chronic disorder.1 Together with daytime hypersomnolence, it is estimated to occur in approximately 14% of men and 5% of women aged 30–70 years.2 OSA is associated with multiple comorbidities (including obesity), reduced quality of life, increased motor vehicle accidents and increased risk of cardiovascular mortality.3, 4, 5, 6 Excess weight is considered to be the most important risk factor for OSA.7, 8 Moreover, the relationship between obesity and OSA appears to be bidirectional.5 Approximately 60–70% of individuals with OSA are overweight,9, 10 and about 58% of moderate to severe OSA cases are attributable to excess weight.8
Weight loss has been shown to reduce OSA severity and improve blood oxygen saturation and sleep architecture parameters, as well as self-reported quality of life.11, 12, 13, 14, 15 The beneficial effects of initial weight loss on OSA severity have been shown to persist over the long-term despite weight regain.13, 16, 17 Current clinical practice guidelines recommend weight loss for adults with excess weight and OSA.18, 19
45Nasal continuous positive airway pressure (CPAP) is considered to be the gold standard treatment for moderate to severe OSA. However, treatment adherence is often suboptimal (46–83% of individuals fail to adhere), and CPAP does not address the underlying pathophysiology (that is, excess weight) for overweight individuals with OSA.20 For individuals with obesity and moderate or severe OSA who cannot or will not use CPAP, treatment options are limited.
Liraglutide, a glucagon-like peptide-1 (GLP-1) analog with a 97% homology to human GLP-1, is approved for the treatment of type 2 diabetes at once-daily doses up to 1.8 mg.21 Liraglutide has recently received marketing approval for weight management at the 3.0 mg dose.22, 23, 24 Liraglutide’s weight loss mechanism in humans is through a reduction in appetite with a subsequent decrease in energy intake, not increased energy expenditure.25
The aim of this 32-week randomized, controlled trial was to investigate whether liraglutide 3.0 mg would reduce the severity of OSA when compared with placebo (both as adjunct to a reduced-calorie diet and increased physical activity) in participants with obesity and moderate or severe OSA who were unwilling or unable to use CPAP therapy. Although both interventions were evaluated in conjunction with lifestyle modifications, they will be abbreviated as ‘liraglutide’ and ‘placebo’ hereafter. Liraglutide’s weight loss efficacy, as well as the relationship between weight loss, sleep apnea end points and sleep/health-related quality of life, were also examined.
Materials and methods
Study overview
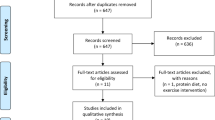
This 32-week randomized, double-blind, placebo-controlled parallel-group trial was conducted between June 2012 and June 2013 at 40 sites in the United States and Canada (Figure 1). The trial was conducted in accordance with the Declaration of Helsinki26 and Good Clinical Practice Guidelines.27 Local institutional review boards approved the trial protocol and all amendments. All participants provided written informed consent.

Trial design. EOT, end of treatment; FU, follow-up.
Participants
The complete list of inclusion/exclusion criteria is provided in Supplementary Table E1. In brief, eligible individuals were men and women aged 18–64 years with a stable body weight (<5% change during the previous 3 months) and body mass index (BMI) of ⩾30 kg m−2. Eligible individuals had to be diagnosed with moderate (apnea–hypopnea index (AHI) 15.0–29.9 events h−1) or severe (AHI ⩾30.0 events h−1) OSA and be unable or unwilling to use CPAP therapy. Individuals with central sleep apnea and/or type 1 or 2 diabetes were excluded from enrollment.
Intervention
Eligible participants were randomly assigned 1:1 in a blinded, centralized manner (using a sponsor-provided telephone- or web-based system) to once-daily subcutaneous liraglutide or placebo. To reduce the likelihood of gastrointestinal symptoms,22, 23, 24 liraglutide was started at the 0.6 mg day−1 dose and escalated in weekly 0.6-mg increments to 3.0 mg (week 4); Figure 1. The 3.0 mg dose was maintained for another 28 weeks. A placebo dose-volume equivalent was used to maintain blinding. In this double-blind trial, the participants, investigators/site personnel and sponsor were blinded to treatment assignments. All participants received counseling on diet and physical activity approximately every 4 weeks during treatment. Based on the individual total energy expenditure estimated by the investigator (using the World Health Organization estimates and an ‘average’ physical activity factor of 1.3),28 each participant was prescribed a daily energy intake 500 kcal below the aforementioned estimate. The diet prescribed approximately 30% of energy from fat, 20% from protein and 50% from carbohydrates. In addition, participants were advised to increase their physical activity to at least 150 min per week. Adherence to lifestyle modifications was encouraged through the distribution of 3-day food diaries (every 5–8 weeks) and pedometers, but was not assessed as an end point.
Outcome measures
The primary efficacy end point was change in AHI (using the American Academy of Sleep Medicine’s recommended 2007 definition, with hypopnea scoring requiring ⩾30% reduction in nasal pressure signal excursions from baseline and ⩾4% desaturation from pre-event baseline)29 from baseline to week 32. Key secondary efficacy end points included changes from baseline to week 32 in OSA severity category, blood oxygen saturation parameters (lowest oxygen saturation, percentage of time with oxygen saturation <90% and oxygen desaturation ⩾4% index), sleep architecture parameters (total sleep time, wake time after sleep onset, proportion of sleep spent in supine position and sleep stage distribution), body weight-related parameters (fasting body weight, proportion of participants losing ⩾5% or >10% of baseline fasting body weight, BMI, waist and neck circumference), glycemic parameters (HbA1c and fasting plasma glucose), vital signs (systolic blood pressure (SBP) and diastolic blood pressure, pulse), fasting lipids (high-density lipoprotein, low-density lipoprotein, very-low-density lipoprotein, and total cholesterol and triglycerides), cardiovascular biomarkers (high-sensitivity C-reactive protein and urinary albumin:creatinine ratio), daytime sleepiness (Epworth Sleepiness Scale) and self-reported quality of life (Functional Outcomes of Sleep Questionnaire (FOSQ) and 36-item Short-Form (SF-36) health status survey). Questionnaire total score can range from 0 to 24, with a lower score indicating a lower propensity for daytime sleepiness.30 FOSQ total score can range from 5 to 20, with a higher score indicating less functional impairment.31 The SF-36 scores can range from 0 to 100, with higher scores indicating a better quality of life.32
Polysomnographic (PSG) assessments (including AHI, blood oxygen saturation parameters, sleep architecture parameters and nocturnal heart rate) were performed during overnight clinic stays at weeks −1 (screening), 12 and 32, and data were scored centrally by Clinilabs (New York, NY, USA). Body weight-related parameters and vital signs were assessed at every visit from week 0 to 32. After randomization, glycemic parameters, fasting lipids, cardiovascular biomarkers and quality of life were assessed at weeks 0, 12 and 32; fasting plasma glucose was also assessed at weeks 4, 8, 20 and 28.
Safety assessments included adverse events (AEs), standard laboratory tests, physical examinations, electrocardiograms and mental health (Patient Health Questionnaire-9 and Columbia Suicide Severity Rating Scale). Specific attention was given to certain types of AEs, including those related to obesity and/or weight loss or side-effects relevant to the GLP-1 receptor agonist drug class (Supplementary Table E2).
Statistical analysis
The sample size was calculated using a two-sided t-test with a significance level of 5%. A sample size of 308 was estimated to provide a ⩾85% power to detect a treatment difference of 6 events h−1 for the primary end point, assuming a s.d. of 17 events h−1 (based on previous publications). The sample size calculation was also based on the assumption that 5% of participants would withdraw before trial completion without any post-baseline data for the primary analysis (see next paragraph) of the primary end point. In fact, 7% of participants withdrew from the trial without providing any post-baseline data for the primary end point (Supplementary Methods and Supplementary Table E3).
The efficacy analyses were performed on the data from the full analysis set, which included all randomized participants. The safety analysis set included all randomized participants who were exposed to ⩾1 dose(s) of trial drug. The last observation carried forward imputation method was pre-specified for primary and secondary analyses. A detailed description of the imputation approach used for the primary end point is provided in the Supplementary Methods. The presented results are based on last observation carried forward imputation unless otherwise noted. The robustness of the primary analysis was investigated by multiple sensitivity analyses using other methods for handling missing data (Supplementary Table E3).
Continuous end points were analyzed using a pre-specified analysis of covariance model with treatment, gender and country as fixed effects and baseline BMI, age and value at baseline as covariates. The null hypothesis was rejected on a 5% level if the two-sided 95% confidence interval (CI) of the treatment difference excluded 0. Pre-specified analyses of categorical end points were performed using logistic regression, with the same fixed effects and covariates as the analysis of covariance. Exploratory analyses of the influence of weight loss on selected end points were conducted post hoc. Post hoc analysis of covariance models also included a weight change (% or kg) covariate and examined its interactions with other effects (for example, OSA severity category at baseline). No adjustment for multiple testing was applied to the secondary analyses and the results should be evaluated accordingly.
Unless otherwise specified, mean changes from baseline are presented as observed means±s.d. (or s.e.) in the main manuscript. Model-estimated mean changes from baseline are presented in the Supplementary Table E4. Safety data were summarized descriptively. Statistical analyses were performed with SAS software, version 9.3 (SAS Institute Inc., Cary, NC, USA).
Results
Trial population
Of the 359 randomized participants, 276 (liraglutide: 134 (74%), placebo: 142 (79%)) completed the trial (Figure 2). A higher proportion of participants withdrew because of AEs in the liraglutide than in the placebo group (12% vs 3%); gastrointestinal AEs most commonly led to trial withdrawal in the liraglutide group. More participants in the placebo group than in the liraglutide group withdrew consent to participate (11% vs 7%).

Trial flow. Participants could be ineligible for trial enrollment because of more than one inclusion/exclusion criterion. *Participants did not receive any trial drug and withdrew from the trial. **Non-compliance with the trial protocol included, but was not limited to, incorrect handling of trial product, non-compliance to visit schedule and dietary advice and non-completion of trial-related questionnaires.
Baseline characteristics of randomized participants were similar between the two treatment groups (Table 1).28, 33 Consistent with the greater prevalence of OSA in males, >70% of trial participants were male. The majority of participants had severe OSA (~67%). The mean baseline AHI of approximately 49 events h−1 revealed the high severity of the disease. Approximately half of the participants with moderate OSA were females, whereas participants with severe OSA were predominantly males (~80%). Generally, participants with severe OSA were heavier, and more had comorbidities than those with moderate OSA.
Polysomnography end points
In both treatment groups, most of the reduction in mean AHI occurred by week 12, with minimal changes thereafter (Figure 3a). At week 32, the mean reduction in AHI with liraglutide was statistically significantly greater compared with placebo (−12.2±1.8 vs −6.1±2.0 events h−1; estimated treatment difference: −6.1 events h−1 (95% CI, −11.0 to −1.2), P=0.015) (Figure 4a and Supplementary Table E4). The robustness of the primary analysis findings was confirmed with several sensitivity analyses (Supplementary Table E3). The treatment effect on AHI did not depend on participants’ gender, baseline BMI, or OSA severity category (P>0.05 for interaction with treatment for all). However, participants with severe OSA at baseline experienced greater mean reduction in AHI in both treatment groups. The majority of participants in both treatment groups did not experience improvement/worsening of their baseline OSA severity category after 32 weeks (Supplementary Figure E1, panel A). However, there was a trend for more liraglutide-treated than placebo-treated participants to no longer meet the diagnostic criteria for OSA (that is, AHI <5 events h−1) (5.4% vs 1.2%, respectively; odds ratio (OR) 4.4 (95% CI, 0.9–21.0), P=0.07) or experience a 50% reduction in their baseline AHI score (31.5% vs 21.7%, respectively, OR 1.6 (95% CI, 1.0–2.7), P=0.05). The changes in oxygen saturation and sleep architecture parameters presented in Figure 4a and Supplementary Table E4 did not differ significantly between the liraglutide and placebo groups. In addition, no notable changes from baseline in sleep stage distribution were observed at week 32 (end point was not analyzed statistically). The change in the proportion of sleep time spent in the supine position after 32 weeks did not differ significantly between treatment groups (P=0.20).

Change in AHI and body weight over 32 weeks of treatment and the relationship between weight loss and change in AHI. (a) Change in AHI during the treatment period. (b) Change in body weight during the treatment period. (c) Relationship between weight loss and change in AHI. (a, b) Data are observed means±s.e. from the full analysis set (FAS). The last observation carried forward (LOCF) value is shown at week 32. In c, data are mean±s.e. from the FAS, LOCF. Weight gain or loss category represents the change in body weight (%) from baseline after 32 weeks of treatment. PSG assessments were performed at weeks –1 (baseline), 12 and 32. For PSG-derived parameters (that is, AHI), participants without any post-baseline PSG assessments were excluded from the analyses. For the body weight parameter, participants without any post-baseline measurements were excluded from the analyses; in addition, participants without baseline measurements were excluded from the change from baseline analyses. N, number of participants; n, number of participants in each weight loss category; WG, weight gain; WL, weight loss.

Changes in PSG, quality of life and cardiometabolic end points after 32 weeks of treatment. (a) Changes in PSG and quality of life end points. (b) Changes in cardiometabolic end points. Forest plots show estimated treatment differences (ETDs)/odds ratios and 95% CIs. Data are from the full analysis set (last observation carried forward (LOCF)). Data at baseline are mean±s.d. For PSG-derived parameters, participants without any post-baseline PSG assessments were excluded from the analyses. For quality of life and cardiometabolic parameters, participants without any post-baseline measurements were excluded from the analyses; in addition, participants without baseline measurements were excluded from the change from baseline analyses. Improvement/worsening refer to the changes from baseline with liraglutide 3.0 mg relative to those with placebo. DBP, diastolic blood pressure; ESS, Epworth Sleepiness Scale; FPG, fasting plasma glucose; I, improvement; ODI, oxygen desaturation index; SpO2, oxygen saturation; WASO, wake time after sleep onset.
Body weight and related end points
Mean body weight decreased continuously in the liraglutide 3.0 mg group over the 32 weeks (Figure 3b). In the placebo group, the mean weight loss curve appeared to plateau around week 12. At week 32, the mean weight loss was significantly greater with liraglutide than with placebo (−5.7±0.4% vs −1.6±0.3%, estimated treatment difference: −4.2%, (95% CI, −5.2 to −3.1%), P<0.0001; Figure 4b and Supplementary Table E4). Greater proportions of liraglutide- versus placebo-treated participants lost either ⩾5% (46.3% vs 18.5%, OR: 3.9 (95% CI, 2.4–6.4), P<0.0001) or >10% (23.4% vs 1.7%, OR: 19.0 (95% CI, 5.7–63.1), P<0.0001) of baseline body weight at week 32. Consistent with the greater weight loss effect, significantly greater reductions in BMI, waist and neck circumference were also observed with liraglutide compared with placebo.
Glycemic control and cardiometabolic parameters
Liraglutide 3.0 mg produced significantly greater reductions from baseline in HbA1c and fasting plasma glucose than placebo at week 32 (Figure 4b and Supplementary Table E4). Liraglutide also significantly reduced SBP compared with placebo, but had no significant effect on diastolic blood pressure. No statistically significant improvements in fasting lipids or the urinary albumin:creatinine ratio were observed with liraglutide compared with placebo. The reduction in high-sensitivity C-reactive protein with liraglutide was of borderline significance versus placebo (P=0.05).
Sleep/health-related quality of life
Significantly greater improvements with liraglutide 3.0 mg than placebo were observed for the ‘general health’ domain of the SF-36 (P=0.0426) and the ‘activity level’ domain of the FOSQ (P=0.015). The remaining total and individual domain scores on the three questionnaires (Epworth Sleepiness Scale (ESS), FOSQ and SF-36) did not differ significantly between groups (Figure 4a and Supplementary Table E4).
Relationship between weight loss and selected sleep-related end points
Treatment assignment, independent of weight loss, did not significantly affect the change from baseline in AHI (P=0.82); that is, the effect of treatment was mediated by weight loss in both groups. In both treatment groups, greater weight loss was significantly associated with greater reduction in AHI (Figure 3c). Similarly, larger proportions of participants tended to improve their OSA severity category and lesser proportions tended to worsen their OSA severity category with greater weight loss (Supplementary Figure E1, panels B and C). In post hoc analysis, the reduction in AHI per 1% (or 1 kg) weight loss depended on baseline AHI, with reductions of 0.7 (0.7), 1.4 (1.3) and 2.8 (2.2) events h−1 for baseline AHI cohorts <30, 30–59 and ⩾60 events h−1, respectively (P<0.001, both groups). Greater weight loss at the end of the trial was also significantly associated with greater end-of-trial improvements in end points related to oxygen saturation, sleep architecture and total scores on Epworth Sleepiness Scale and FOSQ (P<0.01 for all) (Supplementary Figure E2).
Safety
More participants reported AEs with liraglutide (80.1%) than with placebo (69.3%), driven primarily by the greater prevalence of gastrointestinal AEs with liraglutide (mainly nausea). Gastrointestinal AEs were mostly mild or moderate in severity and transient, occurring mainly within the initial 8 weeks of treatment. Most participants experiencing AEs reported events that were either mild (liraglutide: 69.9%, placebo: 57.5%) or moderate (liraglutide: 50.0%, placebo: 36.3%) in severity. No deaths occurred. Identical proportions of participants in both groups (3.4%) reported serious AEs (Table 2). Overall, serious AEs occurred as single cases and there was no notable pattern of distribution with respect to the system organ class. Based on the low number of reported special-focus events in both groups, no clear patterns of distribution or differences between treatments were identified. No events of pancreatitis or medullary thyroid carcinoma were reported. In total, two AEs of cholelithiasis (one serious) were reported with liraglutide and one serious AE of cholecystitis was reported with placebo. Amylase and lipase activity and calcitonin are described in the Supplementary Results.
Similar to previous findings with GLP-1 receptor agonists, a significantly greater increase in mean resting pulse was seen with liraglutide compared with placebo at week 32 (1.6±0.8 vs 0.8±0.8 beats min−1, estimated treatment difference 2.1 beats min−1 (95% CI, 0.3–4.0), P=0.0256). Normal physiological decline in nocturnal heart rate was observed in both treatment groups during the 8- h PSG assessment, but less so with liraglutide (Supplementary Figure E3). Compared with previous findings with liraglutide doses up to 1.8 mg in diabetes, no new safety issues with liraglutide 3.0 mg were identified during clinical laboratory evaluation, physical examinations and electrocardiogram assessment. No notable differences between liraglutide and placebo were observed during mental health evaluations with Patient Health Questionnaire-9 and Columbia Suicide Severity Rating Scale.
Discussion
This trial examined the effect of weight loss on OSA severity in participants with obesity and moderate/severe OSA who were unable or unwilling to use CPAP therapy. After 32 weeks of treatment, a statistically significantly greater reduction in mean AHI was seen with liraglutide 3.0 mg compared with placebo, both adjunctive to diet and exercise counseling. Accompanying the AHI reduction, consistent trends for improvement were also seen with liraglutide versus placebo across oxygen saturation, sleep architecture and sleep/health-related quality of life end points. Improvements in OSA end points were related to the degree of weight loss at the end of the study, with greater improvement seen with greater weight loss. This observation is in line with those from several other randomized, controlled studies in patients with OSA, irrespective of whether weight loss was induced by lifestyle intervention, pharmacotherapy or surgery.11, 12, 14, 34, 35 The effect of weight loss on AHI appeared to be most pronounced in participants with severe OSA at baseline, an observation also previously reported in other weight loss studies in participants with obesity and OSA.11, 13 Although the present trial was not specifically designed to detect treatment differences for shifts in OSA severity categories, greater percentages of participants improved severity category with greater weight loss in both groups. At week 32, weight loss with liraglutide had not plateaued. Thus, it is possible that further weight loss-related improvement in AHI would have been seen with longer liraglutide treatment duration. The precise clinical significance of the 12 events h−1 mean reduction in AHI with liraglutide is unclear, but consistent positive changes in supportive secondary end points related to sleep apnea and cardiovascular risk factors indicate an overall trend toward improvement. Although no significant improvements in supportive sleep apnea end points were seen in this population with mainly severe disease (baseline AHI ~49 events h−1), the statistical/clinical significance of AHI reduction with liraglutide may become more apparent in patients with a less severe form of the disease (that is, mild to moderate). This is a subject for further studies.
Individuals with OSA often have a co-aggregation of other adverse conditions (that is, obesity, metabolic syndrome, dysglycemia/diabetes) that predispose them to increased cardiovascular risk.5 This clustering of risk factors was exemplified in the present trial population; in addition to moderate/severe OSA and obesity, 42% of participants had hypertension, 33% had dyslipidemia and 63% had prediabetes (classification was based on the HbA1c and fasting plasma glucose criteria of the American Diabetes Association 2010 definition)33 at baseline; individuals with diabetes were excluded from the trial. In addition to greater reductions in AHI, participants treated with liraglutide 3.0 mg also experienced significantly greater improvements in other measures of cardiovascular risk, including SBP and blood glucose, both of which are well-described effects of liraglutide treatment in participants with and without diabetes.21, 36, 37 SBP reductions of a similar magnitude to that observed with liraglutide 3.0 mg in this trial (3.4 mm Hg reduction from baseline) have been associated with lower cardiovascular risk/mortality.38, 39 Consistent with observations for the GLP-1 receptor agonist drug class, treatment with liraglutide increased mean resting heart rate by 2 beats min−1 compared with placebo in this trial.40, 41 As participants with OSA are characterized by increased sympathetic tone,42, 43 it is reassuring that in this trial the increase in heart rate was similar or smaller and the reduction in SBP was greater than those previously observed with liraglutide 3.0 mg in participants without OSA.37, 44 The long-term clinical effects of the increase in heart rate have not been established. Results from the cardiovascular outcomes trial with liraglutide in patients with type 2 diabetes mellitus (LEADER) indicate liraglutide treatment is associated with a statistically significant reduction in cardiovascular risk (assessed using a composite outcome (first occurrence of cardiovascular death, non-fatal myocardial infarction or non-fatal stroke during the 3.5–5 year trial period)).45
The safety profile of liraglutide 3.0 mg in this trial was consistent with that previously observed with liraglutide doses up to 1.8 mg in diabetes and with liraglutide 3.0 mg in the other trials within the weight management program.21, 37, 46 No new safety issues were identified. As typically observed with GLP-1 receptor agonists,47, 48 transient gastrointestinal AEs (mainly nausea) occurred more frequently with liraglutide 3.0 mg, but generally subsided after the initial 8 weeks of treatment.
The strengths of the study include its sample size of approximately 360 participants. In addition, meticulous recording of PSG data at specialized sleep sites and the central reading of all PSG results increased data reliability. The study was limited by its 32-week duration, which precluded the achievement of maximum weight loss and observation of effect durability on OSA-related parameters and body weight. Although power was maintained, approximately 23% of participants across both groups withdrew before trial completion. This attrition rate is reasonable given the difficulty of retaining patients in sleep apnea trials (because of the discomfort/burden associated with overnight PSG assessments), and corresponds to that seen in another OSA trial with a large sample size.11 Importantly, the findings for the primary end point were generally consistent across various methods of handling missing data.
Conclusion
In participants with obesity and moderate or severe OSA, 32 weeks of treatment with liraglutide 3.0 mg (combined with lifestyle modification) produced significantly greater reductions in AHI and improved several other measures of cardiovascular risk, including body weight, blood glucose and SBP, when compared with placebo (that is, lifestyle modifications alone). Greater weight loss led to greater improvements in AHI and other sleep-related end points. This study suggests that liraglutide 3.0 mg in conjunction with a diet and exercise regimen may be useful as a weight management component of a comprehensive therapeutic approach for OSA management. Given its range of beneficial pharmacodynamic effects, liraglutide 3.0 mg may also be a useful tool in individuals already on CPAP treatment.
References
Young T, Peppard PE, Gottlieb DJ . Epidemiology of obstructive sleep apnea: a population health perspective. Am J Respir Crit Care Med 2002; 165: 1217–1239.
Peppard PE, Young T, Barnet JH, Palta M, Hagen EW, Hla KM . Increased prevalence of sleep-disordered breathing in adults. Am J Epidemiol 2013; 177: 1006–1014.
Nieto FJ, Young TB, Lind BK, Shahar E, Samet JM, Redline S et al. Association of sleep-disordered breathing, sleep apnea, and hypertension in a large community-based study. Sleep Heart Health Study. JAMA 2000; 283: 1829–1836.
Shamsuzzaman AS, Gersh BJ, Somers VK . Obstructive sleep apnea: implications for cardiac and vascular disease. JAMA 2003; 290: 1906–1914.
Tuomilehto H, Seppa J, Uusitupa M . Obesity and obstructive sleep apnea - clinical significance of weight loss. Sleep Med Rev 2012; 17: 321–329.
Young T, Finn L, Peppard PE, Szklo-Coxe M, Austin D, Nieto FJ et al. Sleep disordered breathing and mortality: eighteen-year follow-up of the Wisconsin sleep cohort. Sleep 2008; 31: 1071–1178.
Young T, Skatrud J, Peppard PE . Risk factors for obstructive sleep apnea in adults. JAMA 2004; 291: 2013–2016.
Young T, Peppard PE, Taheri S . Excess weight and sleep-disordered breathing. J Appl Physiol (1985) 2005; 99: 1592–1599.
Lindberg E, Gislason T . Epidemiology of sleep-related obstructive breathing. Sleep Med Rev 2000; 4: 411–433.
Pillar G, Shehadeh N . Abdominal fat and sleep apnea: the chicken or the egg? Diabetes Care 2008; 31 (Suppl 2): S303–S309.
Foster GD, Borradaile KE, Sanders MH, Millman R, Zammit G, Newman AB et al. A randomized study on the effect of weight loss on obstructive sleep apnea among obese patients with type 2 diabetes: the Sleep AHEAD study. Arch Intern Med 2009; 169: 1619–1626.
Johansson K, Neovius M, Lagerros YT, Harlid R, Rossner S, Granath F et al. Effect of a very low energy diet on moderate and severe obstructive sleep apnoea in obese men: a randomised controlled trial. BMJ 2009; 339: b4609.
Kuna ST, Reboussin DM, Borradaile KE, Sanders MH, Millman RP, Zammit G et al. Long-term effect of weight loss on obstructive sleep apnea severity in obese patients with type 2 diabetes. Sleep 2013; 36: 641–649A.
Tuomilehto HP, Seppa JM, Partinen MM, Peltonen M, Gylling H, Tuomilehto JO et al. Lifestyle intervention with weight reduction: first-line treatment in mild obstructive sleep apnea. Am J Respir Crit Care Med 2009; 179: 320–327.
Yee BJ, Phillips CL, Banerjee D, Caterson I, Hedner JA, Grunstein RR . The effect of sibutramine-assisted weight loss in men with obstructive sleep apnoea. Int J Obes (Lond) 2007; 31: 161–168.
Tuomilehto H, Seppa J, Uusitupa M, Tuomilehto J, Gylling H . Weight reduction and increased physical activity to prevent the progression of obstructive sleep apnea: a 4-year observational postintervention follow-up of a randomized clinical trial. [corrected]. JAMA Intern Med 2013; 173: 929–930.
Tuomilehto H, Seppa J, Uusitupa M, Peltonen M, Martikainen T, Sahlman J et al. The impact of weight reduction in the prevention of the progression of obstructive sleep apnea: an explanatory analysis of a 5-year observational follow-up trial. Sleep Med 2014; 15: 329–335.
Epstein LJ, Kristo D, Strollo PJ Jr, Friedman N, Malhotra A, Patil SP et al. Clinical guideline for the evaluation, management and long-term care of obstructive sleep apnea in adults. J Clin Sleep Med 2009; 5: 263–276.
Qaseem A, Holty JE, Owens DK, Dallas P, Starkey M, Shekelle P . Management of obstructive sleep apnea in adults: a clinical practice guideline from the American College of Physicians. Ann Intern Med 2013; 159: 471–483.
Weaver TE, Grunstein RR . Adherence to continuous positive airway pressure therapy: the challenge to effective treatment. Proc Am Thorac Soc 2008; 5: 173–178.
Blonde L, Russell-Jones D . The safety and efficacy of liraglutide with or without oral antidiabetic drug therapy in type 2 diabetes: an overview of the LEAD 1-5 studies. Diabetes Obes Metab 2009; 11 (Suppl 3): 26–34.
Saxenda (liraglutide), U.S. Prescribing Information. January 2015. Available at http://www.novo-pi.com/saxenda.pdf (accessed 16 July 2015).
Saxenda Canada Product Monograph. Available at http://www.novonordisk.ca/content/dam/Canada/AFFILIATE/www-novonordisk-ca/OurProducts/PDF/Saxenda_PM_English.pdf (accessed 17 July 2015).
Summary of Product Characteristics, Saxenda. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003780/WC500185786.pdf (accessed 16 July 2015).
van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WH . Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int J Obes (Lond) 2013; 38: 784–793.
World Medical Association. Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects. Last amended by the 59th WMA Assembly, Seoul, October 2008. Available at www.wma.net/en/30publications/10policies/b3/ (accessed 16 July 2015).
International Conference on Harmonisation. ICH Harmonised Tripartite Guideline. Good Clinical Practice. 01-May-1996. Available at http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R1_Guideline.pdf (accessed 16 July 2015).
FAO/WHO/UNU. Human energy requirements. Report of a joint FAO/WHO/UNU expert consultation. FAO: food and nutrition technical report series 1. Rome: FAO/WHO/UNU; 2004. Available at http://www.fao.org/docrep/007/y5686e/y5686e00.htm (accessed 16 July 2015).
The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications. Available at http://www.aasmnet.org/scoringmanual/ (accessed 16 July 2015).
Johns MW . A new method for measuring daytime sleepiness: the Epworth sleepiness scale. Sleep 1991; 14: 540–545.
Weaver TE, Laizner AM, Evans LK, Maislin G, Chugh DK, Lyon K et al. An instrument to measure functional status outcomes for disorders of excessive sleepiness. Sleep 1997; 20: 835–843.
Ware JE, Kosinski M, Dewey JE . How to Score Version Two of the SF-36 Health Survey. QualityMetric: Lincoln, RI, USA, 2001.
American Diabetes Association. Standards of medical care in diabetes—2010. Diabetes Care 2010; 33 (Suppl 1): S11–S61.
Dixon JB, Schachter LM, O’Brien PE, Jones K, Grima M, Lambert G et al. Surgical vs conventional therapy for weight loss treatment of obstructive sleep apnea: a randomized controlled trial. JAMA 2012; 308: 1142–1149.
Winslow DH, Bowden CH, DiDonato KP, McCullough PA . A randomized, double-blind, placebo-controlled study of an oral, extended-release formulation of phentermine/topiramate for the treatment of obstructive sleep apnea in obese adults. Sleep 2012; 35: 1529–1539.
Astrup A, Rossner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet 2009; 374: 1606–1616.
Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond) 2012; 36: 843–854.
Grossman E . Blood pressure: the lower, the better: the con side. Diabetes Care 2011; 34 (Suppl 2): S308–S312.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R . Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet 2002; 360: 1903–1913.
Diamant M, Van Gaal L, Stranks S, Northrup J, Cao D, Taylor K et al. Once weekly exenatide compared with insulin glargine titrated to target in patients with type 2 diabetes (DURATION-3): an open-label randomised trial. Lancet 2010; 375: 2234–2243.
Dungan KM, Povedano ST, Forst T, Gonzalez JG, Atisso C, Sealls W et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet 2014; 384: 1349–1357.
Chandra S, Sica AL, Wang J, Lakticova V, Greenberg HE . Respiratory effort-related arousals contribute to sympathetic modulation of heart rate variability. Sleep Breath 2013; 17: 1193–1200.
Somers VK, White DP, Amin R, Abraham WT, Costa F, Culebras A et al. Sleep apnea and cardiovascular disease: an American Heart Association/American College Of Cardiology Foundation Scientific Statement from the American Heart Association Council for High Blood Pressure Research Professional Education Committee, Council on Clinical Cardiology, Stroke Council, and Council On Cardiovascular Nursing. In collaboration with the National Heart, Lung, and Blood Institute National Center on Sleep Disorders Research (National Institutes of Health). Circulation 2008; 118: 1080–1111.
Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Lau D et al. A ransomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med 2015; 373: 11–22.
Novo Nordisk . company announcement, 2016. Available at. https://www.novonordisk.com/bin/getPDF.1991879.pdf (accessed 8 April 2016).
Wadden TA, Hollander P, Klein S, Niswender K, Woo V, Hale PM et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet-induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond) 2013; 37: 1443–1451.
Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 2009; 374: 39–47.
Buse JB, Nauck M, Forst T, Sheu WH, Shenouda SK, Heilmann CR et al. Exenatide once weekly versus liraglutide once daily in patients with type 2 diabetes (DURATION-6): a randomised, open-label study. Lancet 2013; 381: 117–124.
Acknowledgements
This study was sponsored by Novo Nordisk A/S. The authors gratefully acknowledge the important contributions of the study participants, clinical trial personnel and the SCALE Sleep Apnea study group. We thank Trine Kvist, PhD, employed by Novo Nordisk A/S, for performing the statistical analyses and reviewing the manuscript for accuracy. We also thank Irina Nayvelt, PhD, employed by Novo Nordisk Inc., for medical writing assistance and Watermeadow Medical, funded by Novo Nordisk, for editorial and administrative assistance. The SCALE – Sleep Apnoea trial is registered with the clinicaltrials.gov Identifier NCT01557166.
Author contributions
Dr Blackman had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Professor Mignot, Drs Zammit, Claudius, Jensen.
Acquisition, analysis and interpretation of data: all authors.
Critical revision of the manuscript for important intellectual content: all authors.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
Dr Blackman: consultant/advisory board participant for Novo Nordisk, Merck Canada, Valeant Canada, Paladin Labs Inc. Dr Foster: at the time of the study was a scientific advisory board participant for Con Agra Foods, United Health Group and Tate and Lyle, and a consultant to Eisai and Novo Nordisk; currently employed by Weight Watchers International. Dr Zammit: consultant for Acorda, Actelion, Alexza, Arena, Aventis, Biovail, Boehringer-Ingelheim, Cephalon, Elan, Eli Lilly, Evotec, Forest, Glaxo Smith Kline, Jazz, King Pharmaceuticals, Ligand, McNeil, Merck, Neurocrine Biosciences, Organon, Pfizer, Purdue, Renovis, Sanofi-Aventis, Select Comfort, Sepracor, Shire, Somnus, Takeda Pharmaceuticals, Vela, Wyeth; grants/research support from Abbott, Abbvie, Actelion, Ancile, Apnex, Arena, Astra-Zeneca, Aventis, Banyu, Biomarin, BMS, Catalyst, Cephalon Inc., CHDI, Elan, Epix, Eisai, Elminda, Evotec, Forest, Galderma, Genentech, Gilead, Glaxo Smith Kline, Gilead, H. Lundbeck A/S, Janssen, Johnson & Johnson, King, Merck and Co., National Institute of Health (NIH), Neurim, Neurocrine Biosciences, Naurex, Neurim, Neurogen, Novo Nordisk, Organon, Orphan Medical, Otsuka, Pfizer, Predix, Respironics, Saladax, Sanofi-Aventis, Sanofi-Synthelabo, Schering-Plough, Sepracor, Sunovion, Shire, Somaxon, Takeda Pharmaceuticals North America, Targacept, Teva, Thymon, Transcept, UCB Pharma, Ultragenyx, Predix, Vanda, Wyeth-Ayerst Research; honoraria received from Neurocrine Biosciences, King Pharmaceuticals, McNeil, Sanofi-Aventis, Sanofi-Synthelabo, Sepracor, Takeda Pharmaceuticals, Vela Pharmaceuticals, Wyeth-Ayerst Research; ownership interest in Clinilabs, Inc., Clinilabs IPA, Inc., Home Sleep and Respiratory Care, Nationwide Sleep Testing. Dr Rosenberg: received research grant support from Merck, Novo Nordisk, Pfizer, Teva, Jazz Pharmaceuticals, Sunovion Pharmaceuticals and Philips-Respironics. Dr Aronne: advisory board member for Myos Corporation; consultant, speaker, advisor, or received research support from Aspire Bariatrics Inc., Eisai, Ethicon Endo-Surgery Inc., GlaxoSmithKline Consumer Healthcare LP, GI Dynamics, Novo Nordisk, Pfizer, Takeda Pharmaceuticals, Vivus Inc., Zafgen Inc.; ownership interest in Myos Corporation and BMIQ. Dr Wadden: advisory board participant for Orexigen Pharmaceuticals, Inc., Novo Nordisk, Nutrisystem, and Shire Pharmaceuticals, with research support (to the University of Pennsylvania) from the first three companies. Dr Claudius: employee of Novo Nordisk and owns company stock. Dr Jensen: employee of Novo Nordisk and owns company stock. Dr Mignot: advisory board member, consultant for and research support from Jazz Pharmaceuticals; consultant for and research grant from Novo Nordisk; consultant for Reset Pharmaceuticals and Merck; expert witness for FTC.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Blackman, A., Foster, G., Zammit, G. et al. Effect of liraglutide 3.0 mg in individuals with obesity and moderate or severe obstructive sleep apnea: the SCALE Sleep Apnea randomized clinical trial. Int J Obes 40, 1310–1319 (2016). https://doi.org/10.1038/ijo.2016.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2016.52
This article is cited by
-
Cardiovascular Benefits of GLP-1 Receptor Agonists in Patients Living with Obesity or Overweight: A Meta-analysis of Randomized Controlled Trials
American Journal of Cardiovascular Drugs (2024)
-
Efficacy of dapagliflozin in the treatment of HFrEF with obstructive sleep apnea syndrome (DAHOS study): study protocol for a multicentric, prospective, randomized controlled clinical trial
Trials (2023)
-
The future of incretins in the treatment of obesity and non-alcoholic fatty liver disease
Diabetologia (2023)
-
Liraglutide and polycystic ovary syndrome: is it only a matter of body weight?
Journal of Endocrinological Investigation (2023)
