
Abstract
In this closing perspective, the author exposes why targeting a single nutrient like sugar is in his opinion unlikely to be efficient in preventing obesity and metabolic diseases. He defends the proposal that the concept of fructose toxicity is based on major misconceptions of nutritional physiology. He specifically proposes that (1) sugar being a non-essential nutrient does not obligatorily imply that it has no beneficial effect; (2) alterations of blood triglyceride concentration and hepatic glucose production within the normal range may merely reflect adaptations to a fructose-rich diet rather than early markers of diseases; (3) overfeeding is a normal physiological response to exposure to an energy-dense, palatable nutrient rather than the consequence of ‘leptin resistance’; (4) we may presently overemphasize the role of biological regulations and of gene-related heredity when assessing the effects of fructose in particular, and the determinants of obesity in general.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

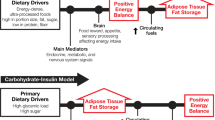
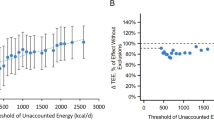
References
Lustig RH, Schmidt LA, Brindis CD . Public health: the toxic truth about sugar. Nature 2012; 482: 27–29.
Eliasen M, Becker U, Gronbaek M, Juel K, Tolstrup JS . Alcohol-attributable and alcohol-preventable mortality in Denmark: an analysis of which intake levels contribute most to alcohol's harmful and beneficial effects. Eur J Epidemiol 2014; 29: 15–26.
Tryon MS, Stanhope KL, Epel ES, Mason AE, Brown R, Medici V et al. Excessive sugar consumption may be a difficult habit to break: a view from the brain and body. J Clin Endocrinol Metab 2015; 100: 2239–2247.
Tappy L, Le KA . Metabolic effects of fructose and the worldwide increase in obesity. Physiol Rev 2010; 90: 23–46.
Lutter M, Nestler EJ . Homeostatic and hedonic signals interact in the regulation of food intake. J Nutr 2009; 139: 629–632.
Breslin PA . An evolutionary perspective on food and human taste. Curr Biol 2013; 23: R409–R418.
Bellisle F, Drewnowski A, Anderson GH, Westerterp-Plantenga M, Martin CK . Sweetness, satiation, and satiety. J Nutr 2012; 142: 1149S–1154S.
Acknowledgements
The author’s work in this area has been supported by grants 320030–135782, 320030–138428, 32003B-156167 and IZ73Z0–152331 from the Swiss National Science Foundation, 11-06 from the Bundes Amt fur Sport BASPO and from Nestle SA, Switzerland and Ajinomoto Inc, Japan. The editorial Aisstance of Mrs E. Tappy is warmly acknowledged. This article is based on a symposium entitled ‘Sweeteners and Health: Findings from Recent Research and their Impact on Obesity and Related Metabolic Conditions’ presented at the 22nd European Congress on Obesity, Prague, on 7 May 2015 with sponsorship from Rippe Lifestyle Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
LT has received lecture fees from Rippe Lifestyle Institute, Nestlé SA and Soremartec. LT has also received grant support from the Swiss National Foundation for Science and Federal Office for Sport BASPO, Switzerland, and serves as an expert witness for the French food security agency ANSES.
Additional information
This article is based on a symposium entitled ‘Sweeteners and Health: Findings from Recent Research and their Impact on Obesity and Related Metabolic Conditions’ presented at the European Congress on Obesity on May 7, 2015 with sponsorship from Rippe Lifestyle Institute.
Rights and permissions
About this article

Cite this article
Tappy, L. What nutritional physiology tells us about diet, sugar and obesity. Int J Obes 40 (Suppl 1), S28–S29 (2016). https://doi.org/10.1038/ijo.2016.11
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2016.11
