
Abstract
Background/Objectives:
Obesity is common following hypothalamic damage due to tumours. Homeostatic and non-homeostatic brain centres control appetite and energy balance but their interaction in the presence of hypothalamic damage remains unknown. We hypothesized that abnormal appetite in obese patients with hypothalamic damage results from aberrant brain processing of food stimuli. We sought to establish differences in activation of brain food motivation and reward neurocircuitry in patients with hypothalamic obesity (HO) compared with patients with hypothalamic damage whose weight had remained stable.
Subjects/Methods:
In a cross-sectional study at a University Clinical Research Centre, we studied 9 patients with HO, 10 age-matched obese controls, 7 patients who remained weight-stable following hypothalamic insult (HWS) and 10 non-obese controls. Functional magnetic resonance imaging was performed in the fasted state, 1 h and 3 h after a test meal, while subjects were presented with images of high-calorie foods, low-calorie foods and non-food objects. Insulin, glucagon-like peptide-1, Peptide YY and ghrelin were measured throughout the experiment, and appetite ratings were recorded.
Results:
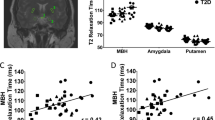
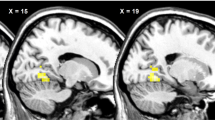
Mean neural activation in the posterior insula and lingual gyrus (brain areas linked to food motivation and reward value of food) in HWS were significantly lower than in the other three groups (P=0.001). A significant negative correlation was found between insulin levels and posterior insula activation (P=0.002).
Conclusions:
Neural pathways associated with food motivation and reward-related behaviour, and the influence of insulin on their activation may be involved in the pathophysiology of HO.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others

References
Daousi C, Dunn AJ, Foy PM, Macfarlane IA, Pinkney JH . Endocrine and neuroanatomic features associated with weight gain and obesity in adult patients with hypothalamic damage. Am J Med 2005; 118: 45–50.
Steele CA, Cuthbertson DJ, Macfarlane IA, Javadpour M, Das KS, Gilkes C et al. Hypothalamic obesity: prevalence, associations and longitudinal trends in weight in a specialist adult neuroendocrine clinic. Eur J Endocrinol 2013; 168: 501–507.
Lawson EA, Holsen LM, Santin M, Meenaghan E, Eddy KT, Becker AE et al. Oxytocin secretion is associated with severity of disordered eating psychopathology and insular cortex hypoactivation in anorexia nervosa. J Clin Endocrinol Metab 2012; 97: E1898–E1908.
Sainte-Rose C, Puget S, Wray A, Zerah M, Grill J, Brauner R et al. Craniopharyngioma: the pendulum of surgical management. Childs Nerv Syst 2005; 21: 691–695.
Holsen LM, Lawson EA, Blum J, Ko E, Makris N, Fazeli PK et al. Food motivation circuitry hypoactivation related to hedonic and nonhedonic aspects of hunger and satiety in women with active anorexia nervosa and weight-restored women with anorexia nervosa. J Psychiatry Neurosci 2012; 37: 322–332.
Tataranni PA, Gautier JF, Chen K, Uecker A, Bandy D, Salbe AD et al. Neuroanatomical correlates of hunger and satiation in humans using positron emission tomography. Proc Natl Acad Sci USA 1999; 96: 4569–4574.
Rosenbaum M, Sy M, Pavlovich K, Leibel RL, Hirsch J . Leptin reverses weight loss-induced changes in regional neural activity responses to visual food stimuli. J Clin Invest 2008; 118: 2583–2591.
Alkan A, Sahin I, Keskin L, Cikim AS, Karakas HM, Sigirci A et al. Diffusion-weighted imaging features of brain in obesity. Magn Reson Imaging 2008; 26: 446–450.
Hinton EC, Parkinson JA, Holland AJ, Arana FS, Roberts AC, Owen AM . Neural contributions to the motivational control of appetite in humans. Eur J Neurosci 2004; 20: 1411–1418.
DelParigi A, Chen K, Salbe AD, Reiman EM, Tataranni PA . Sensory experience of food and obesity: a positron emission tomography study of the brain regions affected by tasting a liquid meal after a prolonged fast. Neuroimage 2005; 24: 436–443.
Small DM, Zatorre RJ, Dagher A, Evans AC, Jones-Gotman M . Changes in brain activity related to eating chocolate: from pleasure to aversion. Brain 2001; 124: 1720–1733.
Gautier JF, Chen K, Salbe AD, Bandy D, Pratley RE, Heiman M et al. Differential brain responses to satiation in obese and lean men. Diabetes 2000; 49: 838–846.
Gautier JF, Del PA, Chen K, Salbe AD, Bandy D, Pratley RE et al. Effect of satiation on brain activity in obese and lean women. Obes Res 2001; 9: 676–684.
Killgore WD, Young AD, Femia LA, Bogorodzki P, Rogowska J, Yurgelun-Todd DA . Cortical and limbic activation during viewing of high- versus low-calorie foods. Neuroimage 2003; 19: 1381–1394.
Rothemund Y, Preuschhof C, Bohner G, Bauknecht HC, Klingebiel R, Flor H et al. Differential activation of the dorsal striatum by high-calorie visual food stimuli in obese individuals. Neuroimage 2007; 37: 410–421.
Simmons WK, Martin A, Barsalou LW . Pictures of appetizing foods activate gustatory cortices for taste and reward. Cereb Cortex 2005; 15: 1602–1608.
Cornier MA, Von Kaenel SS, Bessesen DH, Tregellas JR . Effects of overfeeding on the neuronal response to visual food cues. Am J Clin Nutr 2007; 86: 965–971.
Schur EA, Kleinhans NM, Goldberg J, Buchwald D, Schwartz MW, Maravilla K . Activation in brain energy regulation and reward centers by food cues varies with choice of visual stimulus. Int J Obes (Lond) 2009; 33: 653–661.
Stoeckel LE, Weller RE, Cook EW III, Twieg DB, Knowlton RC, Cox JE . Widespread reward-system activation in obese women in response to pictures of high-calorie foods. Neuroimage 2008; 41: 636–647.
Figlewicz DP, Bennett JL, Aliakbari S, Zavosh A, Sipols AJ . Insulin acts at different CNS sites to decrease acute sucrose intake and sucrose self-administration in rats. Am J Physiol Regul Integr Comp Physiol 2008; 295: R388–R394.
Hallschmid M, Higgs S, Thienel M, Ott V, Lehnert H . Postprandial administration of intranasal insulin intensifies satiety and reduces intake of palatable snacks in women. Diabetes 2012; 61: 782–789.
Takei H, Fujita S, Shirakawa T, Koshikawa N, Kobayashi M . Insulin facilitates repetitive spike firing in rat insular cortex via phosphoinositide 3-kinase but not mitogen activated protein kinase cascade. Neuroscience 2010; 170: 1199–1208.
Schilling TM, Ferreira de Sa DS, Westerhausen R, Strelzyk F, Larra MF, Hallschmid M et al. Intranasal insulin increases regional cerebral blood flow in the insular cortex in men independently of cortisol manipulation. Hum Brain Mapp 2013; 35: 1944–1956.
Daousi C, Macfarlane IA, English PJ, Wilding JP, Patterson M, Dovey TM et al. Is there a role for ghrelin and peptide-YY in the pathogenesis of obesity in adults with acquired structural hypothalamic damage? J Clin Endocrinol Metab 2005; 90: 5025–5030.
Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature 2002; 418: 650–654.
Batterham RL, Cohen MA, Ellis SM, Le Roux CW, Withers DJ, Frost GS et al. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med 2003; 349: 941–948.
Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC . Leptin regulates striatal regions and human eating behaviour. Science 2007; 317: 1355.
Frank S, Heni M, Moss A, von Schnurbein J, Fritsche A, Haring HU, Farooqi S et al. Leptin therapy in a congenital leptin-deficient patient leads to acute and long-term changes in homeostatic, reward, and food-related brain areas. J Clin Endocrinol Metab 2011; 96: E1283–E1287.
Smeets PA, de Graaf C, Stafleu A, van Osch MJ, Nievelstein RA, van der Grond J . Effect of satiety on brain activation during chocolate tasting in men and women. Am J Clin Nutr 2006; 83: 1297–1305.
Acknowledgements
We thank Val Adams and Bill Bimson (MARIARC) for their help with the fMRI procedures; Nicola Williams, Neil Molyneux and Peter Taylor for their help with the purchase, preparation and taking of the food photographs; and Shirley Cooper for her help during the study days. We also thank all patients and healthy volunteers who participated in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
About this article

Cite this article
Steele, C., Powell, J., Kemp, G. et al. Cerebral activations during viewing of food stimuli in adult patients with acquired structural hypothalamic damage: a functional neuroimaging study. Int J Obes 39, 1376–1382 (2015). https://doi.org/10.1038/ijo.2015.82
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2015.82

{kind=link}