
Abstract
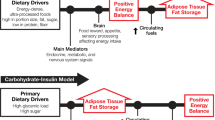
Energy homeostasis is critical for the survival of species. Therefore, multiple and complex mechanisms have evolved to regulate energy intake and expenditure to maintain body weight. For weight maintenance, not only does energy intake have to match energy expenditure, but also macronutrient intake must balance macronutrient oxidation. However, this equilibrium seems to be particularly difficult to achieve in individuals with low fat oxidation, low energy expenditure, low sympathetic activity or low levels of spontaneous physical activity, as in addition to excess energy intake, all of these factors explain the tendency of some people to gain weight. Additionally, large variability in weight change is observed when energy surplus is imposed experimentally or spontaneously. Clearly, the data suggest a strong genetic influence on body weight regulation implying a normal physiology in an ‘obesogenic’ environment. In this study, we also review evidence that carbohydrate balance may represent the potential signal that regulates energy homeostasis by impacting energy intake and body weight. Because of the small storage capacity for carbohydrate and its importance for metabolism in many tissues and organs, carbohydrate balance must be maintained at a given level. This drive for balance may in turn cause increased energy intake when consuming a diet high in fat and low in carbohydrate. If sustained over time, such an increase in energy intake cannot be detected by available methods, but may cause meaningful increases in body weight. The concept of metabolic flexibility and its impact on body weight regulation is also presented.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
References
Ravussin E . Obesity in britain. Rising trend may be due to ‘pathoenvironment’. BMJ 1995; 311: 1569.
Speakman JR . A nonadaptive scenario explaining the genetic predisposition to obesity: the ‘predation release’ hypothesis. Cell Metab 2007; 6: 5–12.
Obesity: preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser 2000; 894: i–xii, 1–253.
Bouchard C . The biological predisposition to obesity: beyond the thrifty genotype scenario. Int J Obes (Lond) 2007; 31: 1337–1339.
Schulz LO, Bennett PH, Ravussin E, Kidd JR, Kidd KK, Esparza J et al. Effects of traditional and western environments on prevalence of type 2 diabetes in Pima Indians in Mexico and the US. Diabetes Care 2006; 29: 1866–1871.
Flegal KM, Carroll MD, Ogden CL, Johnson CL . Prevalence and trends in obesity among US adults, 1999–2000. JAMA 2002; 288: 1723–1727.
Troiano RP, Flegal KM . Overweight children and adolescents: description, epidemiology, and demographics. Pediatrics 1998; 101: 497–504.
Must A, Spadano J, Coakley EH, Field AE, Colditz G, Dietz WH . The disease burden associated with overweight and obesity. JAMA 1999; 282: 1523–1529.
Wolf AM, Colditz GA . Social and economic effects of body weight in the United States. Am J Clin Nutr 1996; 63: 466S–469S.
Alpert SS . Growth, thermogenesis, and hyperphagia. Am J Clin Nutr 1990; 52: 784–792.
Ravussin E, Swinburn BA . Metabolic predictors of obesity: crosssectional versus longitudinal data. Int J Obes Relat Metab Disord 1993; 17: S28–S31.
Ravussin E, Lillioja S, Anderson TE, Christin L, Bogardus C . Determinants of 24-h energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest 1986; 78: 1568–1578.
Weyer C, Snitker S, Rising R, Bogardus C, Ravussin E . Determinants of energy expenditure and fuel utilization in man: effects of body composition, age, sex, ethnicity and glucose tolerance in 916 subjects. Int J Obes Relat Metab Disord 1999; 23: 715–722.
Hill JO, Peters JC . Environmental contributions to the obesity epidemic. Science 1998; 280: 1371–1374.
Hill JO, Wyatt HR, Reed GW, Peters JC . Obesity and the environment: where do we go from here? Science 2003; 299: 853–855.
Zelewski M, Swierczynski J . Comparative studies on lipogenic enzyme activities in the liver of human and some animal species. Comp Biochem Physiol B 1990; 95: 469–472.
Chascione C, Elwyn DH, Davila M, Gil KM, Askanazi J, Kinney JM . Effect of carbohydrate intake on de novo lipogenesis in human adipose tissue. Am J Physiol 1987; 253: E664–E669.
Just B, Messing B, Darmaun D, Rongier M, Camillo E . Comparison of substrate utilization by indirect calorimetry during cyclic and continuous total parenteral nutrition. Am J Clin Nutr 1990; 51: 107–111.
Manji S, Shikora S, McMahon M, Blackburn GL, Bistrian BR . Peritoneal dialysis for acute renal failure: overfeeding resulting from dextrose absorbed during dialysis. Crit Care Med 1990; 18: 29–31.
Acheson KJ, Schutz Y, Bessard T, Flatt JP, Jequier E . Carbohydrate metabolism and de novo lipogenesis in human obesity. Am J Clin Nutr 1987; 45: 78–85.
Acheson KJ, Schutz Y, Bessard T, Anantharaman K, Flatt JP, Jequier E . Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am J Clin Nutr 1988; 48: 240–247.
Snyder WS, Cook NJ, Nasset ES, Karhausen LR, Howells GP, Tipton IH . Report of the task group on reference man. The International Commission on Radiological Protection, no. 23 edn, Pergamon Press: New York, 1974.
Bray GA . Treatment for obesity: a nutrient balance/nutrient partition approach. Nutr Rev 1991; 49: 33–45.
Abbott WG, Howard BV, Christin L, Freymond D, Lillioja S, Boyce VL et al. Short-term energy balance: relationship with protein, carbohydrate, and fat balances. Am J Physiol 1988; 255: E332–E337.
Schutz Y, Flatt JP, Jequier E . Failure of dietary fat intake to promote fat oxidation: a factor favoring the development of obesity. Am J Clin Nutr 1989; 50: 307–314.
Flatt JP, Ravussin E, Acheson KJ, Jequier E . Effects of dietary fat on postprandial substrate oxidation and on carbohydrate and fat balances. J Clin Invest 1985; 76: 1019–1024.
Rising R, Alger S, Boyce V, Seagle H, Ferraro R, Fontvieille AM et al. Food intake measured by an automated food-selection system: relationship to energy expenditure. Am J Clin Nutr 1992; 55: 343–349.
Schutz Y, Tremblay A, Weinsier RL, Nelson KM . Role of fat oxidation in the long-term stabilization of body weight in obese women. Am J Clin Nutr 1992; 55: 670–674.
Hellerstedt WL, Jeffery RW, Murray DM . The association between alcohol intake and adiposity in the general population. Am J Epidemiol 1990; 132: 594–611.
Colditz GA, Giovannucci E, Rimm EB, Stampfer MJ, Rosner B, Speizer FE et al. Alcohol intake in relation to diet and obesity in women and men. Am J Clin Nutr 1991; 54: 49–55.
Shelmet JJ, Reichard GA, Skutches CL, Hoeldtke RD, Owen OE, Boden G . Ethanol causes acute inhibition of carbohydrate, fat, and protein oxidation and insulin resistance. J Clin Invest 1988; 81: 1137–1145.
Frayn KN . Physiological regulation of macronutrient balance. Int J Obes Relat Metab Disord 1995; 19: S4–S10.
Knowler WC, Pettitt DJ, Saad MF, Charles MA, Nelson RG, Howard BV et al. Obesity in the Pima Indians: its magnitude and relationship with diabetes. Am J Clin Nutr 1991; 53: 1543S–1551S.
Ravussin E, Lillioja S, Knowler WC, Christin L, Freymond D, Abbott WG et al. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med 1988; 318: 467–472.
Tataranni PA, Harper IT, Snitker S, Del Parigi A, Vozarova B, Bunt J et al. Body weight gain in free-living Pima Indians: effect of energy intake vs expenditure. Int J Obes Relat Metab Disord 2003; 27: 1578–1583.
Astrup A, Gotzsche PC, van de Werken K, Ranneries C, Toubro S, Raben A et al. Meta-analysis of resting metabolic rate in formerly obese subjects. Am J Clin Nutr 1999; 69: 1117–1122.
Amatruda JM, Statt MC, Welle SL . Total and resting energy expenditure in obese women reduced to ideal body weight. J Clin Invest 1993; 92: 1236–1242.
Weinsier RL, Nelson KM, Hensrud DD, Darnell BE, Hunter GR, Schutz Y . Metabolic predictors of obesity. Contribution of resting energy expenditure, thermic effect of food, and fuel utilization to four-year weight gain of post-obese and never-obese women. J Clin Invest 1995; 95: 980–985.
Zurlo F, Ferraro RT, Fontvielle AM, Rising R, Bogardus C, Ravussin E . Spontaneous physical activity and obesity: cross-sectional and longitudinal studies in Pima Indians. Am J Physiol 1992; 95: E296–E300.
Snitker S, Tataranni PA, Ravussin E . Spontaneous physical activity in a respiratory chamber is correlated to habitual physical activity. Int J Obes Relat Metab Disord 2001; 25: 1481–1486.
Levine JA, Eberhardt NL, Jensen MD . Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science 1999; 283: 212–214.
Johannsen DI, Ravussin E . Spontaneous physical activity: relationship between fidgeting and body weight control. Curr Opin Endocrinol Diabetes Obes 2008; 15: 409–415.
Spraul M, Ravussin E, Fontvieille AM, Rising R, Larson DE, Anderson EA . Reduced sympathetic nervous activity. A potential mechanism predisposing to body weight gain. J Clin Invest 1993; 92: 1730–1735.
Schwartz RS, Jaeger LF, Veith RC . Effect of clonidine on the thermic effect of feeding in humans. Am J Physiol 1988; 254: R90–R94.
Christin L, O’Connell M, Bogardus C, Danforth Jr E, Ravussin E . Norepinephrine turnover and energy expenditure in Pima Indian and white men. Metabolism 1993; 42: 723–729.
Snitker S, Tataranni PA, Ravussin E . Respiratory quotient is inversely associated with muscle sympathetic nerve activity. J Clin Endocrinol Metab 1998; 83: 3977–3979.
Tataranni PA, Young JB, Bogardus C, Ravussin E . A low sympathoadrenal activity is associated with body weight gain and development of central adiposity in Pima Indian men. Obes Res 1997; 5: 341–347.
Astrup A . The sympathetic nervous system as a target for intervention in obesity. Int J Obes Relat Metab Disord 1995; 19 (Suppl 7): S24–S28.
McNeill G, Bruce AC, Ralph A, James WP . Inter-individual differences in fasting nutrient oxidation and the influence of diet composition. Int J Obes 1988; 12: 455–463.
Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol 1990; 259: E650–E657.
Toubro S, Sorensen TI, Hindsberger C, Christensen NJ, Astrup A . Twenty-four-hour respiratory quotient: the role of diet and familial resemblance. J Clin Endocrinol Metab 1998; 83: 2758–2764.
Seidell JC, Muller DC, Sorkin JD, Andres R . Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: the Baltimore Longitudinal Study on Aging. Int J Obes Relat Metab Disord 1992; 16: 667–674.
Astrup A, Buemann B, Christensen NJ, Toubro S . Failure to increase lipid oxidation in response to increasing dietary fat content in formerly obese women. Am J Physiol 1994; 266: E592–E599.
Larson DE, Ferraro RT, Robertson DS, Ravussin E . Energy metabolism in weight-stable postobese individuals. Am J Clin Nutr 1995; 62: 735–739.
Froidevaux F, Schutz Y, Christin L, Jequier E . Energy expenditure in obese women before and during weight loss, after refeeding, and in the weight-relapse period. Am J Clin Nutr 1993; 57: 35–42.
Stock MJ . Gluttony and thermogenesis revisited. Int J Obes Relat Metab Disord 1999; 23: 1105–1117.
Bouchard C, Tremblay A, Despres JP, Nadeau A, Lupien PJ, Theriault G et al. The response to long-term overfeeding in identical twins. N Engl J Med 1990; 322: 1477–1482.
Neumann R . Experimentelle Beitrage zur lehre von dem taglichen Nahrungsbedarf des Menschen unter besonderer Berucksichtigung der notwendigen Eiweissmenge. Arch Hyg 1902; 45: 1–87.
Miller DS, Mumford P, Stock MJ . Gluttony. 2. Thermogenesis in overeating man. Am J Clin Nutr 1967; 20: 1223–1229.
Sims EA, Danforth Jr E, Horton ES, Bray GA, Glennon JA, Salans LB . Endocrine and metabolic effects of experimental obesity in man. Recent Prog Horm Res 1973; 29: 457–496.
Befroy DE, Peterson KF, Dufour S, Mason GF, Rothman DL, Shulman GI . Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. PNAS 2008; 105: 16701–16706.
Lebon V, Dufour S, Petersen KF, Ren J, Jucker BM, Slezak LA et al. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest 2001; 108: 733–737.
Jucker BM, Dufour S, Ren J, Cao X, Previs SF, Underhill B et al. Assessment of mitochondrial energy coupling in vivo by 13C/31P NMR. Proc Natl Acad Sci USA 2000; 97: 6880–6884.
Fernstrom M, Bakkman L, Tonkonogi M, Shabalina IG, Rozhdestvenskaya Z, Mattsson CM et al. Reduced efficiency, but increased fat oxidation, in mitochondria from human skeletal muscle after 24-h ultraendurance exercise. J Appl Physiol 2007; 102: 1844–1849.
Rosenbaum M, Vandenborne K, Goldsmith R, Simoneau JA, Heymsfield S, Joanisse DR et al. Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am J Physiol Regul Integr Comp Physiol 2003; 285: R183–R192.
Coll AP, Farooqi IS, O’Rahilly S . The hormonal control of food intake. Cell 2007; 129: 251–262.
Russek M . Participation of hepatic glucoreceptors in the control of intake of food. Nature 1963; 197: 79–80.
Flatt JP . The difference in the storage capacities for carbohydrate and for fat, and its implications in the regulation of body weight. Ann NY Acad Sci 1987; 499: 104–123.
Hardie DG, Scott JW, Pan DA, Hudson ER . Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 2003; 546: 113–120.
Goldberg GR, Murgatroyd PR, McKenna AP, Heavey PM, Prentice AM . Dietary compensation in response to covert imposition of negative energy balance by removal of fat or carbohydrate. Br J Nutr 1998; 80: 141–147.
Snitker S, Larson DE, Tataranni PA, Ravussin E . Ad libitum food intake in humans after manipulation of glycogen stores. Am J Clin Nutr 1997; 65: 941–946.
Shetty PS, Prentice AM, Goldberg GR, Murgatroyd PR, McKenna AP, Stubbs RJ et al. Alterations in fuel selection and voluntary food intake in response to isoenergetic manipulation of glycogen stores in humans. Am J Clin Nutr 1994; 60: 534–543.
Stubbs RJ, Murgatroyd PR, Goldberg GR, Prentice AM . Carbohydrate balance and the regulation of day-to-day food intake in humans. Am J Clin Nutr 1993; 57: 897–903.
Pannacciulli N, Salbe AD, Ortega E, Venti CA, Bogardus C, Krakoff J . The 24-h carbohydrate oxidation rate in a human respiratory chamber predicts ad libitum food intake. Am J Clin Nutr 2007; 86: 625–632.
Langhans W, Scharrer E . Metabolic control of eating. World Rev Nutr Diet 1992; 70: 1–67.
Eckel RH, Hernandez TL, Bell ML, Weil KM, Shepard TY, Grunwald GK et al. Carbohydrate balance predicts weight and fat gain in adults. Am J Clin Nutr 2006; 83: 803–808.
Galgani JE, Heilbronn LK, Azuma K, Kelley DE, Albu JB, Pi-Sunyer X et al. Metabolic flexibility in response to glucose is not impaired in people with type 2 diabetes after controlling for glucose disposal rate. Diabetes 2008; 57: 841–845.
Ukropcova B, Sereda O, de Jonge L, Bogacka I, Nguyen T, Xie H et al. Family history of diabetes links impaired substrate switching and reduced mitochondrial content in skeletal muscle. Diabetes 2007; 56: 720–727.
Schrauwen P, Westerterp KR . The role of high-fat diets and physical activity in the regulation of body weight. Br J Nutr 2000; 84: 417–427.
Schrauwen P, van Marken Lichtenbelt WD, Saris WH, Westerterp KR . Changes in fat oxidation in response to a high-fat diet. Am J Clin Nutr 1997; 66: 276–282.
Hill JO, Peters JC, Reed GW, Schlundt DG, Sharp T, Greene HL . Nutrient balance in humans: effects of diet composition. Am J Clin Nutr 1991; 54: 10–17.
Smith SR, de Jonge L, Zachwieja JJ, Roy H, Nguyen T, Rood JC et al. Fat and carbohydrate balances during adaptation to a high-fat. Am J Clin Nutr 2000; 72: 450–457.
Acknowledgements
Jose Galgani is supported by a fellowship from the International Nutrition Foundation/Ellison Medical Foundation. Eric Ravussin is supported by grants funded by the National Institutes of Health, U01-AG020478 and RO1-DK60412-06A1.
Author information
Authors and Affiliations
Corresponding author
Additional information
Conflict of interest
The authors have declared no financial interests.
Rights and permissions
About this article
Cite this article
Galgani, J., Ravussin, E. Energy metabolism, fuel selection and body weight regulation. Int J Obes 32 (Suppl 7), S109–S119 (2008). https://doi.org/10.1038/ijo.2008.246
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2008.246
Keywords
This article is cited by
-
Issues related to the assessment of energy balance during short-term over-, under- and refeeding in normal weight men
European Journal of Clinical Nutrition (2023)
-
Splicing factor SRSF1 deficiency in the liver triggers NASH-like pathology and cell death
Nature Communications (2023)
-
Differential disruption on glucose and insulin metabolism in two rat models of diet-induced obesity, based on carbohydrates or lipids
Molecular and Cellular Biochemistry (2023)
-
Reproducibility of the energy metabolism response to an oral glucose tolerance test: influence of a postcalorimetric correction procedure
European Journal of Nutrition (2023)
-
Relationship of BMI with the diet, physical activity and oral hygiene practices amongst the dental students
BMC Oral Health (2022)
