
Abstract
Plant hormones are important molecules which at low concentration can regulate various physiological processes. Mass spectrometry has become a powerful technique for the quantification of multiple classes of plant hormones because of its high sensitivity and selectivity. We developed a new ultrahigh pressure liquid chromatography–full-scan high-definition accurate mass spectrometry method, for simultaneous determination of abscisic acid and four metabolites phaseic acid, dihydrophaseic acid, 7′-hydroxy-abscisic acid and abscisic acid glucose ester, cytokinins zeatin, zeatin riboside, gibberellins (GA1, GA3, GA4 and GA7) and indole-3-acetyl-L-aspartic acid. We measured the amount of plant hormones in the flesh and skin of two processing potato cvs. Sylvana and Russet Burbank stored for up to 30 weeks at 6 °C under ambient air conditions. Herein, we report for the first time that abscisic acid glucose ester seems to accumulate in the skin of potato tubers throughout storage time. The method achieved a lowest limit of detection of 0.22 ng g−1 of dry weight and a limit of quantification of 0.74 ng g−1 dry weight (zeatin riboside), and was able to recover, detect and quantify a total of 12 plant hormones spiked on flesh and skin of potato tubers. In addition, the mass accuracy for all compounds (<5 ppm) was evaluated.
Similar content being viewed by others

Introduction
Plant hormones (PHs) play pivotal roles in plant growth, development and response to biotic and abiotic stresses. Little information has been reported on the class and abundance of plant hormones on postharvest physiology of fresh produce. Abscisic acid (ABA) has been consistently recognized as a tuber dormancy-related plant hormone, while a decline in ABA content is associated with dormancy progression and appears to be a prerequisite for dormancy break; however, there is most likely no particular threshold concentration below which dormancy is broken.1,2 In contrast, gibberellins (GAs) and cytokinins are thought to be involved in the release of dormancy, whereas ABA and ethylene have been associated with the onset and maintenance of potato tuber dormancy.2 Endogenous levels of GA1, GA19 and GA20 have been measured in potato tuber using gas chromatography coupled to mass spectrometry single ion monitoring (GC–MS–SIM) with internal standards; the endogenous concentration of these GAs in tubers exiting dormancy was essentially the same as that of deeply dormant tubers.2 Collectively, it has been suggested that endogenous GAs are not intimately involved in tuber dormancy control, but they play a critical role in subsequent sprout elongation. It has also been suggested that cytokinins are natural dormancy-terminating agents in potato tubers.3 However, the role of gibberellins and cytokinins in dormancy regulation and control of potato tubers remains obscure. The effects reported in most of the experiments done in the past have used exogenous applications of gibberellins (GA3, GA1) and synthetic cytokinins (6-benzylaminopurine) to excised tuber buds and have rarely measured endogenous levels (naturally occurring) of these plant hormones in tuber tissues.2,4,5 In contrast, some of the findings and measurements of their abundances have been made using monoclonal antibodies against specific cytokinins.3,6 In addition, and to the best of our knowledge, high-definition mass accuracy MS techniques have not been used to measure endogenous levels of PHs in potato tubers.
In the past, great efforts had been made on the development of quantification methods for the simultaneous determination of most plant hormones. Liquid chromatography–mass spectrometry (LC–MS) has been considered as the best technique for the analysis of PHs due to its selectivity and sensitivity. A number of methods have been reported using reversed-phase liquid chromatography electrospray ionization (ESI) tandem mass spectrometry MS/MS.7–9 The majority of these methods have been developed by applying multiple reaction monitoring using a triple quadrupole mass spectrometer. These methods have been applied successfully for the simultaneous determination of different classes of PHs, either in positive or negative ionization mode in two separate instrument runs making the time for analysis longer. Few methods have developed combined acquisition in both positive and negative ionization modes on a single instrument run.10,11
Herein, the development and application of a robust, high-throughput sensitive new method for the simultaneous quantification of 12 endogenous PHs in flesh and skins from potato tubers using reversed-phase ultrahigh-pressure liquid chromatography (UHPLC) coupled to high-definition accurate mass quadrupole time of flight mass spectrometry (Q-ToF MS) is described. The method combines the acquisition mode in both positive and negative polarities under full-scan mode, and requires only thirteen minutes per analytical run.
Materials and methods
Plant material
Potato cvs. Sylvana (Greenvale AP Ltd, Cambs, UK) and Russet Burbank (McCain Foods Ltd, Yorks, UK) were used in this study. The selected potato cultivars are categorized as maincrop (‘Russet Burbank’) and early maincrop (‘Sylvana’) according to the British Potato Variety Database (2009). These varieties arrived at Sutton Bridge Crop Storage Research facilities on 10 October 2008 and were passed over a grading line to remove loose soil, rotten, damaged, green and/or undersized (<45 mm) tubers. Sprout suppressants were not applied to the tubers used in the study. On arrival, tubers underwent a controlled cooling regime from 15 °C (arrival temperature), at a rate of 0.5 °C reduction per day at ambient relative humidity, to a holding temperature of 6 °C, in order to minimize chilling stress and allow time for wound healing. After reaching 6 °C, tubers were stored under continuous ambient air for up to 30 weeks. Tubers (n=3) were collected at five sampling points throughout storage: (i) at harvest; (ii) at 6 weeks storage; (iii) at the time of first indication of sprouting (10% of air-stored tubers showing white sprout tissue development12–14); (iv) 4 weeks later; and (v) at 30 weeks storage. Flesh and skin tissues from both cultivars were analyzed in all sampling points; however due to the insufficient amount of skin sample in potato cv. Sylvana at 30 weeks storage, those skin samples were not analyzed at that time point (Figure 2).

Abscisic acid, Abscisic acid-glucose ester, dihydrophaseic and phaseic acid content (ng g−1 DW) in flesh and skin of potato cv. Sylvana tubers stored under ambient air and sampled at five (flesh) or four (skin) time points: after harvest, at 6 weeks storage, at first indication of sprouting (red arrow), 4 weeks later and at 30 weeks storage (n=3). LSD bars for flesh (A) and skin (B) (P<0.05) are shown.
Sample extraction
Freeze-dried potato samples (150 mg) were mixed with 5 mL of extraction solvent comprised of methanol/water/formic acid mixture (75:20:5, v/v/v). Then 50 µL of 400 ng mL−1 solution of internal standard mixture (10 deuterated labeled compounds) was added to the extraction mixture. The mixture was kept at −20 °C overnight in the dark. Solids were separated by centrifugation at 1132g; the remaining residue after centrifugation was re-extracted with 2 mL of the extraction solvent for 30 min at −20 °C. Supernatants of both extractions were reunified into one fraction and passed through a Sep-Pak vac tC18 cartridge previously conditioned with 5 mL of methanol and equilibrated with 5 mL of 1 M formic acid solution. The eluate was first purged with nitrogen at room temperature to evaporate the organic phase and the residue was freeze dried overnight in the dark at −95 °C. It was subsequently reconstituted with 1 mL of 1 M formic acid. The formic acid solution containing all hormones was then loaded into an Oasis MCX cartridge that had been pre-conditioned with 5 mL of methanol and equilibrated with 5 mL of 1 M formic acid solution. The formic acid solution was later discarded and then the cartridge was eluted with 2 mL of methanol and 2 mL of 0.35 M NH4OH in 60% (v/v) methanol. The solution that passed through contained the plant hormones. This solution was first purged with nitrogen at room temperature to evaporate the organic phase and the residue was freeze dried overnight (in the dark at −95 °C). The final dried material was reconstituted with 400 µL of 0.1% formic acid in ultrapure water containing 20 ng of d6-ABA added to evaluate extraction efficiencies. Five microliters of the reconstituted sample was injected into the LC–MS system to quantify the endogenous PHs.
Chemicals and reagents
Methanol and formic acid HPLC grade solvents were purchased from Fisher Scientific (Leics, UK) and used for sample extraction; HPLC grade water was obtained from a Direct-Q3 ultrapure water system from Millipore (Milford, MA, USA). NH4OH (28%–30%) was purchased from Sigma-Aldrich (Dorset, UK). Acetonitrile, formic acid and methanol LC–MS grade for mobile phase preparation were purchased from Fisher Scientific. Sep-Pak® vac tC18 SPE cartridges containing 500 mg of C18 stationary phase bonded on silica gel matrix, and Oasis® MCX cartridges containing 150 mg of sulfonic acid sorbent were purchased from Waters Corporation (Milford, MA, USA).
Deuterated labeled and unlabeled compounds, [2H3]-dihydrozeatin d3-DHZ; [2H3]-dihydrozeatin riboside d3-DHZR; [2H2]-gibberellin A1 d2-GA1; [2H2]-gibberellin A4 d2-GA4; zeatin ±Z; zeatin riboside ±ZR; gibberellin A1 GA1 and gibberellin A4 GA4 were purchased from OlchemIm Ltd (Olomouc, Czech Republic); [2H3]-dihydrophaseic acid (−)-7′,7′,7′-d3 dihydrophaseic acid (DPA); [2H5]-abscisic acid glucose ester (+)-4,5,8′,8′,8′-d5-ABA-GE; [2H3]-phaseic acid (−)-7′,7′,7′,-d3-PA, [2H4]-7′-hydroxy-abscisic acid (±)-5,8′,8′,8′-d4-7′-hydroxy-ABA; [2H4]-abscisic acid (−)-5,8′8′8′-d4-ABA, [2H6]-abscisic acid (±)-3′,5′,5′,7′,7′,7′-d6-ABA; (−)-DPA, (+)-ABA-GE, (−)-PA, (±)-7′-hydroxy-ABA were obtained from the National Research Council of Canada-Plant Biotechnology Institute; (±)-ABA was purchased from Sigma-Aldrich.
LC–ESI–Q-TOF instrument
Chromatographic separation was achieved on an Agilent 1290 Infinity UHPLC system and comprised of the following: a thermostatted column compartment operated at 30 °C, an cooled autosampler, a binary pump with jet weaver V35 mixer. Separation was achieved by a Zorbax Rapid Resolution High Definition Eclipse Plus C18 column (2.1×50 mm 1.8 µm). Compounds were separated at a flow rate of 0.6 mL min−1 using a linear gradient of solvent A (water+0.1% formic acid) and solvent B (acetonitrile+0.1% formic acid) programed to the following profile; 0 min, 4% B+96% A; 1.5 min, 12.6% B; 7.0 min, 26% B; 10 min 40% B and hold for 0.5 min; 10.6 min 100% B and hold for 1 min, finally the column was taken for re-equilibration for 1.35 min (4% B), giving a total run time of 13 min. The injection volume was 5 µL injected with needle wash for 6 s.
Calibration curves were prepared using seven calibration solution levels containing 5, 10, 25, 50, 75, 100 and 150 ng mL−1 each of the 12 unlabeled compounds prepared with the solvent A (water+0.1% formic acid). Each calibration solution level also contained 50 ng mL−1 of 10 corresponding internal standards (deuterated labeled compounds). Five microliters of each calibration solution were injected in triplicate into the UHPLC system. The area underneath the full scan peak corresponding to more abundant accurate m/z of each molecule was used to calculate the relative response of each standard and internal standard for each concentration level. The relative response was equal to the standard peak area divided by the internal standard peak area. Calibration curves for the 12 compounds were generated by plotting the known concentration of the calibration level against the relative response calculated for each calibration level. Quantification analysis was carried out using an Agilent MassHunter Quantitative Analysis Software B.05.00 (Agilent Technologies, Santa Clara, CA), which automatically calculated the final concentration of each compound in the calibration solution, samples and quality control samples.
Full scan MS and MS/MS experiments
Full scan MS experiments were carried out on an Agilent 6540 Ultra High Definition Accurate Mass Q-TOF LC–MS System (Agilent Technologies). Samples, calibration solutions and quality control samples were analyzed using an electrospray ionization source Agilent Dual Jet Stream source in a time segment method. The first time segment comprised of 1.78 min acquisition, acquired in positive mode where the cytokinins zeatin (Z) and zeatin riboside (ZR) were analyzed. The second time segment comprised of 11.22 min acquisition time period, acquired in negative mode to ABA together with its metabolites DPA, abscisic acid glucose ester (ABA-GE), phaseic acid (PA), 7′-hydroxy-abscisic acid (7′-OH-ABA); indole-3-acetyl-l-aspartic acid (IAAsp) and the GAs (GA1, GA3, GA4 and GA7). For both positive and negative modes the following settings were applied: nebulizer gas temperature (N2) 200 °C, at a flow rate of 8 L min−1, sheath gas temperature (N2) 350 °C at a flow rate of 11 L min−1. Capillary voltage ±4000 V, Nozzle voltage ±500 V, fragmentor +175/−165 V. Full-scan data were acquired on the range of 50–1200 m/z, at a acquisition rate of 3 spectra s−1 and data were recorded in centroid mode, using a maximum time between MS spectra of 3 s. Accurate-mass internal mass calibration was performed automatically using a dual-nebulizer ion source combined with an automated calibrant delivery system, which introduced continuously the internal reference ion solution at approximately 50 µL min−1, the reference ion masses were 119.0363 and 1033.9881 m/z in negative mode; 121.0508 and 922.0097 m/z in positive mode. Peak integration was carried out under full-scan conditions using the extracted ion chromatogram (EIC) of the deprotonated or protonated molecule from the total ion chromatogram with 20 ppm mass extraction window. The accurate mass spectrum and retention time for all compounds were used for both confirmation and quantification purposes, while peak areas of EICs were used for quantification.
MS/MS experiments were previously carried out to confirm compound identification; in product ion scan experiments (MS/MS), product ions were produced by collision-induced dissociation of selected precursor ions using targeted MS/MS experiments, with collision energy previously optimized for each compound and ranged from 10–25 eV with an isolation window of 4 m/z (medium) for all compounds (Supplementary Figure S1). Product ion scans were acquired at the mass range of 100–1200 m/z, with an acquisition rate of 3 spectra s−1. The LC-MS system and data acquisition were controlled by an Agilent MassHunter Data Acquisition software B.04.00 (Agilent Technologies).
Results
Development of a high-definition accurate MS method
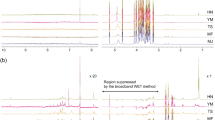
The first step of the analysis for all 12 PHs was to record full-scan spectral data chromatograms. The exact mass to charge ratio (m/z) of the molecules was extracted from the raw data or total ion chromatogram generating an EIC (Figure 1); and the retention time and accurate mass of each compound (standard and internal standard) were obtained. The chromatograms shown on Figure 1 are both EICs, which are free of noise as each peak depicts only the area underneath the accurate mass of each targeted compound where it is most found. The accurate masses of the molecular ions in observed protonated molecule (M+H)+ and deprotonated molecule (M–H)− for positive and negative ionization modes respectively were used for both confirmation and quantification purposes. The chromatograms obtained (EICs) were processed through the quantitative analysis software, which provided the mass accuracy values (difference between measured mass and calculated exact or theoretical mass divided by exact mass and expressed in ppm). Accurate mass measurements were obtained (Table 1) and the errors obtained for most compounds were well below the established accuracy threshold of 5 ppm for unambiguous identification of the 12 PHs and their corresponding deuterated forms. Further unambiguous confirmation of the identity of the PHs was carried out using targeted MS/MS experiments under collision-induced dissociation conditions. The product ion spectra obtained were then compared to match with the standard solution samples (Supplementary Fig. S1) and with data from the published literature obtained with the multiple reaction monitoring method using a triple quadrupole instrument.

(a) UHPLC Q-TOF MS overlaid EICs of a mixture of 14 authentic plant hormone standards, section 1 refers to the first time segment which was acquired in positive ionization mode and section 2 to the second time segment which was acquired in negative ionization mode. (b) Overlaid extracted ion chromatograms of skin tissue extract from potato sample. Compounds are as follows: 0.87, trans-zeatin; 0.98, cis-zeatin; 1.49, trans-zeatin riboside; 1.63, cis-zeatin riboside; 2.06, dihydrophaseic acid; 2.41, indole-3-acetyl-l-aspartic acid, 3.03, gibberellin 3; 3.14, gibberellin 1; 3.39, abscisic acid glucose ester; 3.49, phaseic acid; 4.19, 7′-hydroxy-abscisic acid; 5.50, abscisic acid; 8.77, gibberellin 7; 8.97, gibberellin 4.
Validation of the analytical method
The linearity of the method was evaluated over the range 5–150 ng mL−1 using standard solutions mixture containing 5, 10, 25, 50, 75, 100 and 150 ng mL−1 of each of the 12 unlabeled PHs or their respective metabolites and 50 ng mL−1 of each of their corresponding deuterated forms. Calibration curves for each of the 12 compounds were generated automatically by the quantitative analysis software; briefly the relative response of each compound was plotted against the known concentration level. Linear relationships and good correlation coefficients (R2>0.9996) were obtained for all of the 12 compounds (Table 2).
Instrumental detection limits or limits of detection (LODs) and limits of quantification (LOQs) for each compound were determined based on the signal-to-noise ratio (S/N) and calculated using peak-to-peak algorithm type, where LOD is three times S/N and LOQ is 10 times S/N, the LODs and LOQs obtained were on the range of 0.22–1.50 ng mL−1 and 0.74–5.00 ng mL−1, respectively (Table 2).
The precision of the analytical method was determined by assaying three calibration level points; at 5, 25 and 50 ng mL−1 of each of the 12 PHs on three consecutive times within one day (reproducibility) and with four consecutive days with three replicates each day (repeatability). The relative standard deviations for intra- and inter-day precision were well below 10% for all compounds suggesting good reproducibility and repeatability of the method for the 12 PHs tested (Supplementary Information and Table 2).
To evaluate the accuracy of the method on a tissue sample matrix, assays were carried out on a spiked potato sample. As for the intra- and inter-day precision assays, three spiked levels were tested; low concentration level (5 ng mL−1), medium concentration level (25 ng mL−1) and high concentration level (50 ng mL−1). The % recoveries were then evaluated by comparing the amount of each compound recovered with the amount added, taking into account the endogenous hormones levels in the potato sample previously determined. Briefly recoveries (%)=(amount measured–endogenous amount)/spiked amount×100. The mean recoveries for all 12 spiked PHs were between the ranges 87% and 107% (Table 3). In addition, extraction efficiencies using the Sep-Pak tC18 and Oasis MCX cartridges were also evaluated by adding an external standard (d6-ABA) for each spiked level of the potato samples; the extraction recoveries were well >95%. These results suggest that the new method developed was precise, accurate and sensitive enough for the quantification of 12 plant hormones.
Endogenous content of ABA, PA, ABA-GE and DPA in different tissues of potato tubers
The endogenous ABA content in skin tissue of cv. Sylvana varied from 24 ng g−1 dry weight (DW) at harvest to less than 1 ng g−1 DW at the end of 12 weeks storage corresponding to first indication of sprouting (Figure 2). These levels were slightly different for cv. Russet Burbank which varied from 24 ng g−1 DW at 6 weeks storage to 14 ng g−1 DW at 14 weeks of storage at time of first indication of sprouting (Figure 3). For both cultivars, no significant differences were observed in the content of ABA between flesh tissue compared to skin tissue throughout storage (P<0.05).

Abscisic acid, Abscisic acid-glucose ester, dihydrophaseic and phaseic acid content (ng g−1 DW) in flesh and skin of potato cv. Russet Burbank tubers stored under ambient air and sampled at five time points: after harvest, at 6 weeks storage, at first indication of sprouting (red arrow), 4 weeks later and at 30 weeks storage (n=3). LSD bars for flesh (A), skin (B) and flesh and skin (C) (P<0.05) are shown.
At harvest the endogenous PA content was 118 and 161 ng g−1 DW in skin tissues of cvs. Sylvana and Russet Burbank, respectively; the content decreased dramatically to undetectable levels in both cultivars, and this was more pronounced in the skin tissues of cv. Sylvana than in cv. Russet Burbank. In contrast, the decrease in endogenous PA was more pronounced in the flesh of cv. Russet Burbank than in the skin tissue; and the decline was also accompanied by a concomitant decrease in the content of DPA; these ranged from 539 ng g−1 DW to 125 ng g−1 DW. This decrease was also observed for cv. Sylvana where the endogenous concentration of DPA decreased from 1600 ng g−1 DW to approximately 46 ng g−1 DW during storage without there being a significant difference between flesh and skin tissues (P<0.05).
The endogenous content of ABA-GE in cv. Sylvana appeared to increase throughout storage, showing concentrations of 25 ng g−1 DW in flesh tissue and at 37 ng g−1 DW in skin tissue at harvest and an average of 60 ng g−1 DW at the end of 30 weeks storage. In contrast, the ABA-GE levels in skin tissue of cv. Russet Burbank increased from 21 ng g−1 DW at harvest to 633 ng g−1 DW after 30 weeks of storage. To the best of our knowledge, ABA-GE has never been reported in potato tubers; moreover, it has never been related to tuber dormancy.
Discussion
In order to assess the temporal flux of endogenous plant hormones in potato tubers throughout storage, we have developed a full-scan MS method, and tested its performance to quantify a total of 12 plant hormones in flesh and skin tissues from two different potato cvs. Sylvana and Russet Burbank stored for a maximum of 30 weeks at 6 °C under ambient air conditions.
Among the 12 PHs described here, only ZR, ABA, PA, ABA-GE and DPA were detected in flesh and skin of potato tubers, while Z, ABA-7′-hydroxyl, indole-3-acetyl-l-aspartic acid, gibberellins GA3, GA1, GA7 and GA4 were undetectable maybe due to the LOD. However, the LOD described here (for example, for GA1, was 0.55 ng g−1 DW and LOQ 1.84 ng g−1 DW), was sufficient to detect and measure GA1 levels previously reported to be around 1 ng g−1 of fresh weight at 243 days of postharvest storage, measured using GC–MS–SIM.2 The levels of ZR were well below the limit of quantification (1.58 ng mL−1) and consequently were not reported.
ABA acid has been recognized as a tuber dormancy-related plant hormone, yet in this study, no significant differences were observed in the content of ABA either in flesh and skin tissue of both cultivars studied, rather a gradual decrease was observed as the storage time increased supporting the suggestion that ABA may be linked to potato tuber dormancy progression.15–17 The ABA levels reported (∼50 ng g−1 fresh weight), are consistent with those reported by other authors17 since after harvest all periderm tissues of cv. Russet Burbank had slightly higher ABA content than parenchyma tissues.17 The ABA levels at first indication of sprouting in both cultivars here described varied from undetectable levels to 14 ng g−1 DW and no evidence for a minimum level at the start of sprouting could be established; these results support the statement that there is most likely no particular threshold concentration below which dormancy is broken1 and the role of ABA in potato tuber dormancy progression is still unclear.
It has been hypothesized that once formed, ABA is metabolized primarily by oxidation on the 8′ methyl group to form the unstable intermediate 8′-hydroxy-ABA which at the time spontaneously rearranges to form PA that is then reduced to form DPA.18 Our results are consistent with this, as we observed a dramatic decrease in ABA accompanied with a decline in PA and DPA; the latter being the major ABA metabolite to accumulate in tubers of both cultivars, supporting the principal ABA catabolism in potato tubers proceeds through PA to DPA.19,20
In this study, the skin tissue of cv. Russet Burbank potato tubers seemed to accumulate more ABA-GE than cv. Sylvana throughout storage. At harvest, cv. Russet Burbank had almost threefold less DPA than cv. Sylvana and this may result from a possible inhibition of ABA to be converted into DPA by abscisic acid 8′-hydroxylase. The skin tissue of both cultivars appears to accumulate ABA-GE as the storage time increases. This behavior is more marked in the case of cv. Russet Burbank, suggesting that conjugation with glucose rather than hydroxylation was preferred over catabolic pathway and this could potentially be related to water loss stress and sprouting as this was significantly reduced between 16 and 30 weeks of storage for cv. Russet Burbank (data not shown). Moreover, ABA-GE levels have been also reported to increase during dehydration or water stress and during specific seed development and germination stages.10,21,22 Although ABA conjugates had been thought to be physiologically inactive and accumulate in vacuoles during ageing,23 it has also been proposed that ABA-GE may be involved in long distance transport.24,25 In addition, levels of ABA-GE do not always vary in parallel to the change in ABA levels, suggesting that conjugation may be regulated differently in particular tissues and under specific conditions.26 It is important to note that only generic peel and flesh tissue was evaluated so that buds for example were not independently sampled for PHs.
To conclude, the newly developed full-scan Q-TOF MS method is both reliable and allows accurate quantification of 12 PHs. The validation criteria of accuracy, precision and sensitivity were acceptable and comparable with the LC–MS/MS (multiple reaction monitoring) mode. To the best of our knowledge, this is the first time that a full-scan high-definition accurate mass method has been developed for the simultaneous quantification of PHs. In addition, this is the first report that measures ABA-GE in potato tubers. The key advantages of this new method are: firstly, all compounds are analyzed on a single instrument run, using both positive and negative ionization modes and secondly, all compounds are analyzed under high-resolution full-scan mode, which allowed accurate mass measurements for unambiguous identification of all compounds. In terms of mass accuracy, our hybrid ultrahigh-definition accurate mass Q-TOF LC MS system obtained mass errors data well below the established 5 ppm mass accuracy threshold; the method described here should be helpful in quantification analysis of PHs in roots, tubers, fruits and vegetables.
References
Biemelt S, Hajirezaei M, Hentschel E, Sonnewald U . Comparative analysis of abscisic acid content and starch degradation during storage of tubers harvested from different potato varieties. Potato Res 2000; 43: 371–382.
Suttle JC . Physiological regulation of potato tuber dormancy. Am J Potato Res 2004; 81: 253–262.
Suttle JC, Banowetz GM . Changes in cis-zeatin and cis-zeatin riboside levels and biological activity during potato tuber dormancy. Physiol Plantarum 2000; 109: 68–74.
Hartmann A, Senning M, Hedden P, Sonnewald U, Sonnewald S . Reactivation of meristem activity and sprout growth in potato tubers require both cytokinins and gibberellin. Plant Physiol 2011; 155: 776–796.
Rentzsch S, Podzimska D, Voegele A et al. Dose- and tissue-specific interaction of monoterpenes with the gibberellin-mediated release of potato tuber bud dormancy, sprout growth and induction of α-amylases and β-amylases. Planta 2012; 235: 137–151.
Turnbull CGN, Hanke DE . The control of bud dormancy in potato tubers. Measurement of the seasonal pattern of changing concentrations of zeatin-cytokinins. Planta 1985; 165: 366–376.
Izumi Y, Okazawa A, Bamba T, Kobayashi A, Fukusaki E . Development of a method for comprehensive and quantitative analysis of plant hormones by highly sensitive nanoflow liquid chromatography–electrospray ionization–ion trap mass spectrometry. Anal Chim Acta 2009; 648: 215–225.
Alarcón Flores MI, Romero-Gonzalez R, Frenich AG, Vidal JLM . Quechers-based extraction procedure for multifamily analysis of phytohormones in vegetables by UHPLC–MS/MS. J Sep Sci 2011; 34: 1517–1524.
Li Y, Wei F, Dong X, Peng J, Liu S, Chen H . Simultaneous analysis of multiple endogenous plant hormones in leaf tissue of oilseed rape by solid-phase extraction coupled with high-performance liquid chromatography–electrospray ionization tandem mass spectrometry. Phytochem Anal 2011; 22: 442–449.
Chiwocha SDS, Abrams SR, Ambrose SJ et al. A method for profiling classes of plant hormones and their metabolites using liquid chromatography–electrospray ionization tandem mass spectrometry: an analysis of hormones regulation or thermodormancy of lettuce (Lactuca sativa L.) seeds. Plant J 2003; 35: 405–417.
Pan X, Welti R, Wang X . Simultaneous quantification of major phytohormones and related compounds in crude plant extracts by liquid chromatography–electrospray tandem mass spectrometry. Phytochemistry 2008; 69: 1773–1781.
Foukaraki SG, Chope GA, Terry LA . Ethylene exposure after dormancy break is as effective as continuous ethylene to control sprout growth in some UK-grown potato cultivars. Acta Hort 2012; 934: 1175–1181.
Foukaraki SG, Chope GA, Terry LA . 1-MCP Application before continuous ethylene storage supresseus sugar accumulation in the UK-grown potato cultivar ‘Marfona’. Acta Hort 2012; 945: 291–296.
Foukaraki SG, Cools K, Chope GA, Terry LA . Effect of the transition between ethylene and air storage on postharvest quality in six UK-grown potato cultivars. J Hort Sci Biotech 2014; 89: 599–606.
Suttle JC . Postharvest changes in endogenous ABA levels and ABA metabolism in relation to dormancy in potato tubers. Physiol Plantarum 1995; 95: 233–240.
Destefano-Beltran L, Kauber D, Huckle L, Suttle JC . Effects of postharvest storage and dormancy status on ABA content, metabolism and expression of genes involved in ABA biosynthesis and metabolism in potato tuber tissues. Plant Mol Biol 2006; 61: 687–697.
Lulai EC, Suttle JC, Pederson SM . Regulatory involvement of abscisic acid in potato tuber wound-healing. J Exp Bot 2008; 59: 1175–1186.
Krochko JE, Abrams GD, Loewen MK, Abrams SR, Cutler AJ . (+)-abscisic acid 8′-hydroxylase is a cytochrome P450 monooxygenase. Plant Physiol 1998; 118: 849–860.
Saito S, Hirai N, Matsumoto C et al. Arabidopsis CYP707As encode (+)-Abscisic acid 8′-Hydroxylase, a key enzyme in the oxidative catabolism of abscisic acid. Plant Physiol 2004; 134: 1439–1449.
Suttle JC, Lulai EC, Huckle LL, Neubauer JD . Wounding of potato tubers induce increases in ABA biosynthesis and catabolism and alters expression of ABA metabolic genes. J Plant Physiol 2013; 170: 560–566.
Boyer GL, Zeevaart JAD . Isolation and quantitation of β-D-glucopyranosyl abscisate from leaves of xanthium and spinach. Plant Physiol 1982; 70: 227–231.
Hocher V, Sotta B, Maldiney R, Miginiac E . Changes in abscisic acid and its β-d-glucopyranosyl ester levels during tomato (Lycopersicon esculentum Mill.) seed development. Plant Cell Rep 1991; 10: 444–447.
Bray EA, Zeevaart JAD . The compartmentation of abscisic acid and β-d-glucopyranosyl abscisate in mesophyll cells. Plant Physiol 1985; 79: 719–722.
Sauter A, Hartung W . The contribution of internode and mesocotyl tissues to root-to-shoot signalling of abscisic acid. J Exp Bot 2002; 53: 297–302.
Wilkinson S, Davies WJ . ABA-based chemical signalling: the co-ordination of responses to stress in plants. Plant Cell Environ 2002; 25: 195–210.
Nambara E, Marion-Poll A . Abscisic acid biosynthesis and catabolism. Annu Rev Plant Biol 2005; 56: 165–185.
Acknowledgements
The authors would like to thanks the Potato Council Ltd. for partial financial support of this research (Project R412) and Sutton Bridge Crop Storage Research for use of their storage facilities during experiments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Ordaz-Ortiz, J., Foukaraki, S. & Terry, L. Assessing temporal flux of plant hormones in stored processing potatoes using high definition accurate mass spectrometry. Hortic Res 2, 15002 (2015). https://doi.org/10.1038/hortres.2015.2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hortres.2015.2
This article is cited by
-
Effects of seed tuber size on dormancy and growth characteristics in potato double cropping
Horticulture, Environment, and Biotechnology (2023)
-
Untargeted Metabolomics Analytical Strategy Based on Liquid Chromatography/Electrospray Ionization Linear Ion Trap Quadrupole/Orbitrap Mass Spectrometry for Discovering New Polyphenol Metabolites in Human Biofluids after Acute Ingestion of Vaccinium myrtillus Berry Supplement
Journal of the American Society for Mass Spectrometry (2019)
-
Morphological characteristics, anatomical structure, and gene expression: novel insights into gibberellin biosynthesis and perception during carrot growth and development
Horticulture Research (2015)
