
Abstract
Pelizaeus-Merzbacher disease (PMD; MIM #312080) is a rare X-linked recessive disorder. A male neonate presented with severe respiratory distress that required tracheostomy. After the appearance of nystagmus, PMD was suspected as a diagnosis for the patient, and a missense mutation, p.Phe51Val, was identified in PLP1, the gene responsible for PMD. PMD can be a differential diagnosis in a male neonate presenting severe respiratory distress.
Similar content being viewed by others

Pelizaeus-Merzbacher disease (PMD; MIM #312080) is a rare X-linked recessive disorder caused by an abnormal myelin formation defect and subsequent cell loss in the white matter.1 PMD occurs in ~1/50,000 live male births worldwide.2 During infancy or early childhood, PMD patients typically manifest hypotonia and nystagmus followed by severe spasticity, ataxia and cerebellar dysfunction. Most patients exhibit hypomyelination, which can be detected by brain magnetic resonance imaging.3 A final diagnosis can be made through genetic testing for the proteolipid protein 1 gene (PLP1), which encodes PLP1 and DM20. Phenotypic severities of PMD depend on the clinical subtypes of PLP1 abnormalities, deletions/duplications of the entire region and various nucleotide alterations. Greater than half of PMD patients harbor a duplication of the PLP1 gene and exhibit the classical type. In comparison, missense mutations of PLP1 lead to more severe clinical manifestations, including stridor as a respiratory distress.4–6 Here we describe a PMD patient associated with respiratory distress during the neonatal period (Figures 1 and 2).

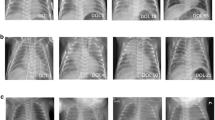
Magnetic resonance imaging (MRI) of the patient. At 2 months of age, T1-weighted cerebral MRI of the patient reveals no signal contrast between white matter and gray matter (a), and T2-weighted MRI exhibits diffuse high signal intensity at the white matter (b). Both images suggest the delay of myelination. These T1/T2-weighted MRI images (c, d) were obtained at approximately the same time at 8 months and confirm the hypomyelination of the patient. No cerebral atrophy is noted.

Sequence chromatogram detected the PLP1 point mutation in exon 2. The patient harbors the missense mutation c.151T>G, which changes Phe to Val at the 51st amino residue (a, b; arrow). His mother was heterozygous at this position and a carrier of the mutation (c; arrow).
A 2-year-old boy had been spontaneously delivered at 38 weeks of gestation. His birth weight was 2,884 g, and his Apgar score was 9/9 at 1 and 5 min, respectively. The parents were non-consanguineous, and the mother was healthy. The family history was unremarkable. Due to generalized hypotonia and stridor, oxygen was supplied until the 11th day. Laryngomalacia was revealed through bronchoscopy and contrast-enhanced computed tomography. He continued to have stridor and soon developed severe respiratory distress. He required intubation on day 33, and tracheostomy was subsequently performed on the 43rd day. Pendular nystagmus was noted on the 50th day. At this time, PMD was listed as a potential candidate diagnosis. Auditory brain-stem response examined by 105 dB only revealed the first wave (Supplementary Figure 1). Brain magnetic resonance imaging examination was performed, and high intensity was detected only in the crus posterus capsulae internae by T2-weighted images, suggesting a hypomyelination pattern. The same radiological findings were detected again at 8 months. After genetic counseling and obtaining informed consent from his parents, genomic DNA was prepared from his family members. Copy numbers of the patient were analyzed and found to be normal.6 Then, nucleotide sequences of all exons and the promotor regions of the PLP1 gene were analyzed by standard Sanger method.7 Finally, NM_000533.4(PLP1): c.151T>G [p.Phe51Val] was detected. His mother was a carrier of this variant. The identified missense mutation, p.Phe51Val, is located on the highly conserved residue among the different species in the extracellular region. Prediction scores of this variant indicated high pathogenicity (0.997, probably damaging by PolyPhen-2; Supplementary Figure 3). From these findings, this patient was diagnosed with the connatal type of PMD. At present, he exhibits good response with smiling and eye contact; however, his head is not controlled yet. Neurological examination revealed rigidity and spasticity due to exaggerated deep tendon reflex and Babinski signs.
Generally, clinical diagnosis of PMD is straightforward if all signs of pyramidal, extrapyramidal and cerebellar tracts are evident. However, diagnosis is often a challenge in early stage of infancy. In this case, nystagmus served as a clue for the differential diagnosis of PMD.5 The patient with a severe, connatal type of PMD exhibited severe respiratory distress requiring tracheostomy. Clinical diagnosis of PMD was first suspected when nystagmus was noted at 50 days. This finding is often observed. The replacement of an amino acid that is highly conserved among species tends to cause a more severe phenotype compared with PLP1 duplication.2,4,6 Phe51Val has been reported previously; however, clinical information related to this mutation was first described in this report.4
In conclusion, patients with a severe form of PMD could present with respiratory distress as an initial symptom during the neonatal period. Even in the absence of nystagmus, we should consider PMD as a differential diagnosis for a male infant with stridor and hypotonia.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Seitelberger F. Neuropathology and genetics of Pelizaeus-Merzbacher disease. Brain Pathol 1995; 5: 267–273.
Numata Y, Gotoh L, Iwaki A, Kurosawa K, Takanashi J, Deguchi K et al. Epidemiological, clinical, and genetic landscapes of hypomyelinating leukodystrophies. J Neurol 2014; 261: 752–758.
Sumida K, Inoue K, Takanashi J, Sasaki M, Watanabe K, Suzuki M et al. The magnetic resonance imaging spectrum of Pelizaeus-Merzbacher disease: a multicenter study of 19 patients. Brain Dev 2016; 38: 571–580.
Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet 2000; 8: 837–845.
Madry J, Hoffman-Zacharska D, Krolicki L, Jakucinski M, Friedman A. PLP1 gene duplication as a cause of the classic form of Pelizaeus-Merzbacher disease—case report. Neurol Neurochir Pol 2010; 44: 511–515.
Inoue K, Osaka H, Sugiyama N, Kawanishi C, Onishi H, Nezu A et al. A duplicated PLP gene causing Pelizaeus-Merzbacher disease detected by comparative multiplex PCR. Am J Hum Genet 1996; 59: 32–39.
Osaka H, Kawanishi C, Inoue K, Onishi H, Kobayashi T, Sugiyama N et al. Pelizaeus-Merzbacher disease: three novel mutations and implication for locus heterogeneity. Ann Neurol 1999; 45: 59–64.
Data Citations
Osaka, Hitoshi HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.1761 (2018)
Acknowledgements
We thank Narumi Ohmika and Shio Aoki for technical support. This work was supported by JSPS KAKENHI Grant Number 15552768 for HO.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplemental Information for this article can be found on the Human Genome Variation website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ueda, A., Shimbo, H., Yada, Y. et al. Pelizaeus-Merzbacher disease can be a differential diagnosis in males presenting with severe neonatal respiratory distress and hypotonia. Hum Genome Var 5, 18013 (2018). https://doi.org/10.1038/hgv.2018.13
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2018.13
