
Abstract
Nuclear receptor subfamily 5, group A, member 1 (NR5A1) is a nuclear receptor involved in gonadal and adrenal development. We identified a novel C-terminally truncating NR5A1 mutation, p.Leu423Trpfs*7, in dizygotic twins with 46,XY disorders of sex development. Our results highlight the functional importance of C-terminal region of NR5A1 and indicate that NR5A1 mutations can be associated with intrafamilial phenotypic variations, progressive testicular dysfunction, hypogonadotropic hypogonadism, and borderline adrenal dysfunction.
Similar content being viewed by others

Nuclear receptor subfamily 5, group A, member 1 (NR5A1; alias SF-1 and Ad4BP) is a nuclear receptor involved in the development of gonads and adrenal glands.1,2 NR5A1 consists of 461 amino acids and contains DNA binding and ligand-binding domains.1,2 The ligand-binding domain is located in the carboxyl (C)-terminal region of the protein and harbors the activation function-2 (AF-2) domain, which mediates the interaction between NR5A1 and its cofactors.1 Although NR5A1 is known as an orphan receptor, recent studies have suggested that phosphatidylinositol (3,4,5)-trisphosphate (PIP3) may be a ligand of NR5A1.3 Blind et al.3 showed that three loop structures in the ligand-binding domain, which are designated as L2–3, L6–7, and L11–12, are essential for the binding of NR5A1 to PIP3.
To date, more than 40 heterozygous mutations in NR5A1 (NM_004959.4) have been identified in patients with 46,XY disorders of sex development (DSD).2 A small percentage of mutation-positive patients exhibited adrenal insufficiency, indicating that during development, testes are more vulnerable to NR5A1 dysfunction than adrenal glands.1,2 Previously reported NR5A1 mutations include five nucleotide alterations in the C-terminal region, namely, p.Val424del, p.Arg427Alafs*139, p.Leu437Gln, p.Leu437Thrfs*57, and p.Glu445*.4–8 Of these, p.Arg427Alafs*139, p.Leu437Thrfs*57, and p.Glu445* remove the L11–12 loop structure and the AF-2 domain, whereas p.Val424del and p.Leu437Gln affect amino acids adjacent to L11–12 (Supplementary Figure S1). In vitro analysis confirmed that p.Val424del and p.Leu437Gln had low-transactivating activities.4,6 Five individuals carrying these C-terminal mutations invariably manifested 46,XY DSD without adrenal insufficiency (Supplementary Figure S1). These findings indicate that the C-terminal region of NR5A1 contains a component essential for protein function in the developing testes. However, considering the limited number of previous reports, further studies are necessary to determine the phenotypic consequences of mutations in the C-terminal region of NR5A1.
Here, we report a hitherto unreported C-terminal truncating mutation in NR5A1 identified in dizygotic male twins. This study was approved by the Institutional Review Board Committee at the National Center for Child Health and Development. Written informed consent was obtained from the patients’ parents. The twins (patients 1 and 2) were born to nonconsanguineous Japanese parents at 38 weeks of gestation after an uncomplicated pregnancy and delivery. At birth, both patients manifested hypomasculinized male-type external genitalia characterized by a micropenis, hypospadias, a bifid scrotum, and undescended testes (Table 1). The patients were otherwise healthy and had no skin pigmentation. They had 46,XY karyotype. Ultrasonography detected bilateral testes in the upper part of scrotum but no Müllerian derivatives. Blood hormone examinations at birth revealed normal testosterone values in patients 1 and 2. However, examinations at two months of age showed compromised testosterone production in both patients; testosterone responses to human chorionic gonadotropin stimulation were blunted in patient 1 and completely absent in patient 2 (Table 1). In addition, patient 2 manifested low-gonadotropin levels at two months of age. Elevated blood levels of adrenocorticotropic hormone in patient 2 and blunted cortisol responses to adrenocorticotropic hormone stimulation in patient 1 suggested subnormal glucocorticoid production. Thus, patients 1 and 2 were diagnosed as having testicular dysfunction and borderline adrenal dysfunction.
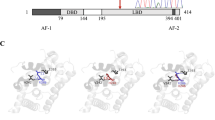
We performed molecular analysis of NR5A1 in patients 1 and 2 and their parents. Genomic DNA samples were extracted from peripheral leukocytes. The entire coding regions of NR5A1 were PCR-amplified and sequenced. The primer sequences are available upon request. Through this analysis we identified a hitherto unreported heterozygous frameshift mutation (c.1267delC, p.Leu423Trpfs*7) in patients 1 and 2 (Figure 1a,b). The mutation was located within the last exon of NR5A1 and satisfied the requirement to escape nonsense-mediated mRNA decay.9 Hence, this mutation was predicted to encode a C-terminally truncated protein lacking L11–12 and the AF-2 domain (Figure 1a). Crystal structure analysis using the PyMol Molecular Graphics System (http://www.pymol.org) suggested that the mutation altered the protein structure of the PIP3 binding site (Figure 1c). The mutation was shared by the phenotypically normal mother of the twins. Next, we performed microsatellite analysis of 13 loci on various chromosomes for the patients and their parents. The results indicated that patients 1 and 2 are dizygotic twins (Supplementary Table S1).

The NR5A1 mutation identified in this study. (a) Schematic representation of wildtype and mutant NR5A1/NR5A1. The black and white boxes in the upper panel indicate the coding and non-coding regions, respectively. The orange and blue boxes in the lower panel indicate the DNA binding and ligand-binding domains, respectively. The activation function-2 (AF-2) domain and three loop structures (L2–3, L6–7, and L11–12) are indicated by yellow and red boxes, respectively. The p.Leu423Trpfs*7 mutation deletes L11–12 and the AF-2 domain and adds six aberrant amino acids (green box). (b) Chromatograms of the c.1267delC mutation. Arrows indicate the mutated nucleotides. (c) Three-dimensional models of NR5A1 and its putative ligand PIP3. Residues in three loop structures (L2–3, L6–7, and L11–12) and H11 mediating the binding between NR5A1 and PIP3 are shown in red. Aberrant amino acids in the mutant protein are shown in green. NR5A1, nuclear receptor subfamily 5, group A, member 1; PIP3, phosphatidylinositol (3,4,5)-trisphosphate.
The aforementioned results indicate that loss of the 38 amino acids at the C-terminal end of NR5A1 weakens the transactivating activity in the fetal testis. Our findings, in conjunction with data of previously reported for five C-terminal mutations,4–8 highlight the functional importance of the C-terminal region of NR5A1 (Supplementary Figure S1). It appears that L11–12 and the AF-2 domain in this region are critical for NR5A1 function because L11–12 has been implicated in PIP3 binding and the AF-2 domain likely mediates the interaction between NR5A1 and other proteins.3 In this regard, the apparently normal phenotype of the mother of patients 1 and 2 is consistent with previous findings that heterozygous NR5A1 mutations usually permit fertility in females, although such mutations can cause decreased ovarian reserve and early menopause.2
Phenotypic analysis of patients 1 and 2 provided several notable findings. First, patient 2 had more severe micropenis and testosterone deficiency than patient 1. These findings are consistent with prior observations that identical NR5A1 mutations can be associated with broad phenotypic variations.8,10,11 Indeed, C-terminal mutations of NR5A1 have been shown to cause 46,XY DSD of various clinical severities without any genotype–phenotype correlations (Supplementary Figure S1).4–8 Our case provides an additional example of the intrafamilial phenotypic variation of NR5A1 abnormalities. Since patients 1 and 2 are dizygotic twins, some genetic factors specific to patient 2 may have enhanced testicular dysfunction in this individual. Second, hormonal evaluation of patient 2 at two months of age showed complete testosterone deficiency, although this individual had partially masculinized external genitalia indicative of moderate androgen exposure at around eight to twelve weeks gestation. Such discrepancy between genital appearance and hormone data were also observed in other male patients with NR5A1 mutations5,6,11–14 These data imply that NR5A1 mutations contribute to progressive testicular dysfunction around birth. Consistent with this, an NR5A1 mutation was identified in a patient with testicular regression syndrome, an extremely rare condition in which 46,XY patients manifest male-type external genitalia in the absence of gonads.10 Additionally, progressive deterioration of testosterone deficiency was reported in a boy carrying a NR5A1 mutation.15 Third, patient 2 manifested low-gonadotropin levels, indicative of hypogonadotropic hypogonadism. In this regard, p.Leu437Gln, one of the five previously reported C-terminal mutations, caused partial hypogonadotropic hypogonadism in addition to testicular dysfunction.6 Gonadotropin deficiency was also reported in pituitary-specific Nr5a1 knockout mice.16 Thus, hypogonadotropic hypogonadism might have contributed to poor testosterone production in patient 2, although we should follow up with this individual until puberty to clarify whether he has gonadotropin deficiency. Finally, hormonal examinations of patients 1 and 2 indicated mildly compromised glucocorticoid production. Thus far, obvious adrenal insufficiency has been documented only in a small fraction of patients with 46,XY DSD due to NR5A1 heterozygous mutations.2 Notably, none of the five previously reported patients with C-terminal mutations manifested adrenal insufficiency.4–8 Our data suggest that mild glucocorticoid deficiency can be hidden in patients with various types of NR5A1 mutations.
In conclusion, we identified a hitherto unreported NR5A1 frameshift mutation in a dizygotic twin pair. Our results highlight the functional importance of the C-terminal region of NR5A1. Furthermore, the results of this study indicate that NR5A1 mutations, including those in the C-terminal region, can be associated with intrafamilial phenotypic variations, progressive testicular dysfunction, hypogonadotropic hypogonadism, and borderline adrenal dysfunction.
References
References
Lin L, Achermann JC . Steroidogenic factor-1 (SF-1, Ad4BP, NR5A1) and disorders of testis development. Sex Dev 2008; 2: 200–209.
Suntharalingham JP, Buonodore F, Duncan AJ, Achermann JC . DAX-1 (NR0B1) and steroidogenic factor-1 (SF-1, NR5A1) in human disease. Best Pract Res Clin Endocrinol Metab 2015; 29: 607–619.
Blind RD, Sablin EP, Kuchenbecker KM, Chiu HJ, Deacon AM, Das D et al. The signaling phospholipid PIP3 creates a new interaction surface on the nuclear receptor SF-1. Proc Natl Acad Sci USA 2014; 111: 15054–15059.
Suwanai AS, Ishii T, Haruna H, Yamataka A, Narumi S, Fukuzawa R et al. A report of two novel NR5A1 mutation families: possible clinical phenotype of psychiatric symptoms of anxiety and/or depression. Clin Endocrinol 2013; 78: 957–965.
Köhler B, Lin L, Ferraz-de-Souza B, Wieacker P, Heidemann P, Schröder V et al. Five novel mutations in steroidogenic factor 1 (SF1, NR5A1) in 46,XY patients with severe underandrogenization but without adrenal insufficiency. Hum Mutat 2008; 29: 59–64.
Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, Albanese A et al. Heterozygous missense mutation in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab 2007; 92: 991–999.
Sıklar Z, Berberoğlu M, Ceylaner S, Çamtosun E, Kocaay P, Göllü G et al. A novel heterozygous mutation in steroidogenic factor-1 in pubertal virilization of a 46,XY female adolescent. J Pediatr Adolesc Gynecol 2014; 27: 98–101.
Woo KH, Cheon B, Kim JH, Cho J, Kim GH, Yoo HW et al. Novel heterozygous mutations of NR5A1 and their functional characteristics in patients with 46,XY disorders of sex development without adrenal insufficiency. Horm Res Paediatr 2015; 84: 116–123.
Kuzmiak HA, Maquat LE . Applying nonsense-mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med 2006; 12: 306–316.
Philibert P, Zenaty D, Lin L, Soskin S, Audran F, Léger J et al. Mutational analysis of steroidogenic factor 1 (NR5a1) in 24 boys with bilateral anorchia: a French collaborative study. Hum Reprod 2007; 22: 3255–3261.
Warman DM, Costanzo M, Marino R, Berensztein E, Galeano J, Ramirez PC et al. Three new SF-1 (NR5A1) gene mutations in two unrelated families with multiple affected members: within-family variability in 46,XY subjects and low ovarian reserve in fertile 46,XX subjects. Horm Res Paediatr 2011; 75: 70–77.
Bertelloni S, Dati E, Baldinotti F, Toschi B, Marrocco G, Sessa MR et al. NR5A1 gene mutations: clinical, endocrine and genetic features in two girls with 46,XY disorders of sex development. Horm Res Paediatr 2014; 81: 104–108.
Mazen I, Abdel-Hamid M, Mekkawy M, Bignon-Topalovic J, Boudjenah R, El Gammal M et al. Identification of NR5A1 mutations and possible digenic inheritance in 46,XY gonadal dysgenesis. Sex Dev 2016; 10: 147–157.
Fabbri HC, Ribeiro de Andrade JG, Maciel-Guerra AT, Guerra-Júnior G, de Mello MP . NR5A1 loss-of-function mutations lead to 46,XY partial gonadal dysgenesis phenotype: report of three novel mutations. Sex Dev 2016; 10: 191–199.
Allali S, Muller JB, Brauner R, Lourenço D, Boudjenah R, Karageorgou V et al. Mutation analysis of NR5A1 encoding steroidogenic factor 1 in 77 patients with 46,XY disorders of sex development (DSD) including hypospadias. PLoS ONE 2011; 6: e24117.
Zhao L, Bakke M, Parker KL . Pituitary-specific knockout of steroidogenic factor 1. Mol Cell Endocrinol 2001; 185: 27–32.
Data Citations
Fukami, Maki HGV Database (2017) http://dx.doi.org/10.6084/m9.figshare.hgv.955
Acknowledgements
This study was supported by Grants-in-Aid from the Japan Society for the Promotion of Science; and by Grants from the Ministry of Health, Labor and Welfare, the Japan Agency for Medical Research and Development, the National Center for Child Health and Development, and the Takeda foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website .
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Hattori, A., Zukeran, H., Igarashi, M. et al. A novel C-terminal truncating NR5A1 mutation in dizygotic twins. Hum Genome Var 4, 17008 (2017). https://doi.org/10.1038/hgv.2017.8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2017.8
