
Abstract
Waardenburg syndrome type 1 (WS1) is a rare autosomal dominant disorder characterized by hair hypopigmentation, abnormal iris pigmentation, and congenital hearing loss. WS1 is caused by mutations in paired box gene 3 (PAX3). We identified a novel PAX3 mutation (c.1107 C>G, p.Ser369Arg) in a Japanese WS1 patient showing abnormal right iris pigmentation, right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism.
Similar content being viewed by others


Mutations in paired box gene 3 (PAX3, NM_181457.3.) are responsible for Waardenburg syndrome type 1 (WS1; OMIM 193500),1,2 which is clinically characterized by hair hypopigmentation, abnormal iris pigmentation, and congenital hearing loss. Its prevalence is estimated to be 1/42,000. Furthermore, it is responsible for 1–3% of total congenital deafness.3 Diagnosis of WS1 is established by clinical findings in most individuals. Five major white forelock and hair hypopigmentation are one pair, dsytopia canthorum and W index>1.95 are one pair. (• congenital hearing loss • white forelock, hair hypopigmentation • abnormal iris pigmentation • dystopia canthorum, W index>0.95 • affected first-degree relative) and minor diagnostic criteria for WS have been proposed by the Waardenburg Consortium.4 An individual must have two major criteria or one major plus two minor criteria to be diagnosed with WS.4,5 PAX3 is one of the nine human PAX genes encoding DNA-binding transcription factors.6 PAX3 has 10 exons and encodes a paired box by exons 2–4 and a homeobox by exons 5 and 6.6
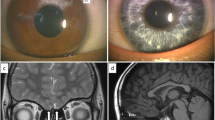
The patient was a 10-month-old Japanese boy born at 33 weeks of gestation with a (very low) birth weight of 1,250 g. He was referred to our hospital because of an incomplete left cleft lip. Body weight at the time of the first visit was 6,700 g at 10 months of age. Although his physique was petite, his nutritional status was good. The patient showed pigmentation abnormalities of the right iris, ocular hypertelorism (W index: 2.00), right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism. He was diagnosed with WS1 on the basis of clinical findings and the criteria proposed by the Waardenburg Consortium.4 According to the parents, his family members, including the proband and mother, showed clinical features consistent with WS1 (Figure 1). The proband is indicated by an arrow (Figure 1a:21). His mother’s hair was speckled with grey. However, she showed no other symptoms (Figure 1a:20). His mother’s aunt showed a lateral iris pigmentation abnormality (Figure 1a:14), and his mother’s elder sister showed a lateral iris pigmentation abnormality and bilateral congenital hearing loss (Figure 1a:19).

(a) Family pedigree of the WS1 patient. Squares indicate males and circles indicate females. Shading in these symbols corresponds to symptoms as follows: upper left quadrant, ocular hypertelorism; lower left, abnormal iris pigmentation; upper right, hearing loss; and lower right, synophrys. (b) Abnormal pigmentation of the right iris of the patient. (c) Ocular hypertelorism (W index: 2.00) of the patient. (d) Incomplete left cleft lip of the patient.
Surgery to repair the cleft lip was performed at 14 months, at a weight of 6,900 g. The preoperative assessment was not problematic.
His parents wanted the patient to undergo a genetic test for PAX3. For the genetic diagnosis, blood samples were obtained from the patient after informed consent was obtained from the parents. Genomic DNA was extracted from the blood samples using a standard protocol. All 10 exons and exon–intron junctions of the PAX3 gene were amplified from genomic DNA by PCR using primers designed with primer software (Genetyx software, Genetyx, Shibuya, Japan). The primers were designed to include an intron of 50 bp surrounding the exon. The sequencing was performed using forward and reverse primers. All the primer sequences are available on request. Sequencing was performed with a BigDye Terminator v3.1 Cycle Sequencing Kit and a 3500×l Genetic Analyzer capillary sequencer (Life Technologies, Carlsbad, CA, USA). Mutation analysis revealed a novel missense mutation (c.1107 C>G, p.Ser369Arg) in exon 7 (Figure 2a).

(a) Sequence chromatograms of the WS1 patient and a control. I: A heterozygous change, c.1107C>G, was identified in the affected boy. II: Healthy controls are wild-type at this position. (b) Schematic diagram of the PAX3 gene. The arrow indicates the mutated gene. The reported missense mutation identified in the WS1 patient is presented. (c) Cross-species multiple alignment of PAX3 protein sequences, showing evolutionary conservation of the altered amino acid, Ser369.
The mutation was predicted to be pathogenic using PolyPhen-2 (probably damaging, Hum Var 0.958, Hum Div 0.776).7 MutationTaster indicated that the mutation was a disease-causing mutation (model: simple_aae, prob: 0.999980746176575).8 Parental analysis was refused. The mutated Ser369 residue is evolutionarily conserved from fish to humans (Figure 2b). Ser369 resides in a highly conserved transactivation domain that interacts with DNA (Figure 2c). These symptoms are believed to arise due to the mutation, which prevents the formation of a covalent disulfide bond (S–S bond), resulting in protein instability.
The variant p.Ser369Arg is located in exon 7. To date, other WS-related mutations have been reported in this exon.9–12 Most of the reported mutations in PAX3 are localized in exons 2–6. Deletions comprise two whole gene deletions and two intragenic deletions of exon 7, and exons 8 and 9.12,13 There is no correlation between the mutation type (missense, nonsense, or deletion) and severity of the phenotype.13 A few mutations of PAX3 have been analyzed for their functional consequences.1,2,13 In addition, functional studies are required to assess the exact molecular mechanisms induced by the c.1107C>G mutation. Cleft lip and palate are low-penetrance features of WS1 and have been found in 2.8% of the reported cases.14
In conclusion, we identified a novel PAX3 mutation (c.1107 C>G, p.Ser369Arg) in a Japanese boy showing pigmentation abnormality in the right iris, right-sided congenital hearing loss, synophrys, incomplete left cleft lip, and cryptorchidism due to WS1. This finding further broadens the existing knowledge of mutations of the PAX3 gene in WS1 patients, enabling clinicians and genetic counsellors to offer better clinical services. WS1 pathogenesis should be taken into account when treating WS1 infants with a cleft lip.
References
References
Baldwin CT, Hoth CF, Amos JA, da-Silva EO, Milunsky A . An exonic mutation in the HuP2 paired domain gene causes Waardenburg’s syndrome. Nature 1992; 355: 637–638.
Hoth CF, Milunsky A, Lipsky N, Sheffer R, Clarren SK, Baldwin CT . Mutation in the paired domain of the human PAX3 gene cause Klein-Waardenburg syndrome(WS-III) as well as Waardenburg syndrome Type-I(WS-I). Am J Hum Genet 1993; 52: 455–462.
Read AP, Newton VE . Waardenburg syndrome. J Med Genet 1997; 34: 656–665.
Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH Jr, Beighton P et al. Waardenburg Syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am J Hum Genet 1992; 50: 902–913.
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK . Waardenburg syndrome. Int J Dermatol 1999; 38: 656–663.
Birrane G, Soni A, Ladias JAA . Structual basis for DNA recognition by the human PAX3 homeodomain. Biochemistry 2009; 48: 1148–1155.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249.
Schwarz JM, Cooper DN, Schuelke M, Seelow D . MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014; 11: 361–362.
Baldwin CT, Hoth CF, Macina RA, Milunsky A . Mutations in PAX3 that cause Waardenburg syndrome type I: Ten new mutations and review of the literature. Am J Med Genet A 1995; 58: 115–122.
Carey ML, Friedman TB, Asher JH Jr, Innis JW . Septo-optic dysplasia and WS1 in the proband of a WS1 family segregating for a novel mutation in PAX3 exon 7. J Med Genet 1998; 35: 248–250.
Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N et al. Review and update of mutations causing Waardenburg syndrome. Human Mutation 2010; 31: 391–406.
Jalilian N, Tabatabaiefar MA, Farhadi M, Bahrami T, Noori-Daloii MR . A novel mutation in the PAX3 gene causes Waardenburg syndrome type I in an Iranian family. Int J Pediatr Otorhinolaryngol 2015; 79: 1736–1740.
Wildhardt G, Zirn B, Graul-Neumann LM, Wechtenbruch J, Suckfüll M, Buske A et al. Spectrum of novel mutations found in Waardenburg syndrome type1 and 2: Implications for molecular genetic diagnosis. BMJ Open 2013; 3: e001917.
Tamayo ML, Gelvez N, Rodriguez M, Florez S, Varon C, Medina D et al. Screening program for Waardenburg syndrome in Colombia: Clinical definition and phenotypic variability. Am J Med Genet A 2008; 146A: 1026–1031.
Data Citations
Yoshida, Yu HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.783
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yoshida, Y., Doi, R., Adachi, K. et al. A novel PAX3 mutation in a Japanese boy with Waardenburg syndrome type 1. Hum Genome Var 3, 16005 (2016). https://doi.org/10.1038/hgv.2016.5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.5
This article is cited by
-
Association of IL-4 with pachychoroid neovasculopathy
Scientific Reports (2023)
