
Abstract
Progressive pseudorheumatoid dysplasia (PPD) is a rare disease caused by mutations in the gene for Wnt1-inducible signaling pathway protein 3 (WISP3). Here, we report the clinical and radiographic manifestations of two Chinese PPD patients. We performed whole-exome sequencing for one patient and sequenced the WISP3 for the other. Three WISP3 mutations (c.396T>G, c.721T>G and c.679dup) were identified; the two missense mutations were novel. Our study expanded the WISP3 mutation spectrum.
Similar content being viewed by others

Progressive pseudorheumatoid dysplasia (PPD; MIM 208230) is an autosomal recessive skeletal dysplasia characterized by the predominant involvement of articular cartilage with progressive stiffness and enlargement of joints in the absence of inflammation. PPD shares similar skeletal manifestations with mucopolysaccharidosis, rheumatoid arthritis and ankylosing spondylitis.1,2 Early development of PPD in patients is normal; however, joint contractures develop in the hands in early childhood and spread to the knees, hips, other large joints, and the spine. Laboratory assessments for rheumatoid factor and inflammation are always negative. A lack of response to anti-rheumatic drugs is characteristic.3 Adult patients are generally severely handicapped due to sustained cartilage loss and pernicious skeletal changes.
PPD is caused by Wnt1-inducible signaling protein 3 (WISP3) mutations. WISP3, located on chromosome 6q22, encodes a connective tissue growth factor involved in cell growth and differentiation.4–6 The expression of WISP3 is vital for human cartilage homeostasis and bone growth, and participates in controlling aggrecan and the cartilage-specific protein collagen II in chondrocytes.7,8 To date, 55 WISP3 mutations have been reported globally, 12 of which were from Chinese patients. In this report, we present the clinical manifestations and radiographic features of two unrelated Chinese PPD patients in whom we found two novel and one recurrent WISP3 mutations.
All participants provided informed consent. The study was approved by the Ethics Committee, Drum Tower Hospital, Nanjing, China and was in accordance with the principles of the Helsinki Declaration.
Patient 1 was a 27-year-old female who presented with multiple joint swelling and hip pain. Her birth history was unremarkable. There was no family history of PPD or other skeletal diseases. The joint swelling began at the age of 12 in the proximal interphalangeal joints and then developed in the elbows and knees without distinct pain or limited motion in the joints. By the age of 14, she noticed limb paresis with an unstable gait. She had difficulty walking, occasional joint pain, and increasing stiffness and swelling of the limb joints and spine. She underwent bilateral total hip joint replacement at our center because she lost her mobility completely after a fall at age 20. On physical examination, she had normal intelligence and face. Her height was 150 cm (−2.8 s.d.). A short trunk without kyphosis, flexural elbows and a limited range of movement in the wrists were noted. She suffered from progressive joint pain, especially in the back. Laboratory examinations, including assessments of inflammation markers and rheumatoid factor, were within the normal range.
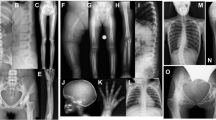
Her X-ray imaging revealed defective ossification of the anterior portions of the upper and lower end plates and slightly flattened thoracic vertebrae (Figure 1a,b). There was a mild S-shaped deformity of the tibiae without other abnormalities. Narrow joint spaces, irregular densities and cyst-like structures in the femoral head and neck indicated marked precocious osteoarthrosis (Figure 1c). The articular surfaces of the knee were flat and irregular, and the joint space had disappeared (Figure 1d). The ends of the short tubular bones were broad, which was most pronounced in the distal ends of the proximal phalanges. The joint spaces of the carpal bones were narrow (Figure 1e). The interphalangeal joints appeared swollen (Supplementary Figure S1a).

Radiographic features of the two patients with progressive pseudorheumatoid dysplasia. (a–e) Patient 1 at age 15. (f–i) Patient 2 at age 17. (a, b) Thoracolumbar spine. Platyspondyly with anterior breaking of the vertebral bodies. (c) Pelvis. Narrow joint spaces and subluxation of the hip joints and irregular densities and cyst-like structures in the femoral head and neck. (d) Knee. The articular surface was flat and irregular, and the joint space was narrow. (e) Hand. Enlargement of the interphalangeal and metacarpal epiphyses. (f) Chest. Decreased heights of the vertebrae. (g–i) Large joints of the lower extremities showed narrow joint spaces.
Patient 2 was a 17-year-old male referred to us because of short stature. His parents were cousins and were healthy with no evidence of arthritis. His limb muscle weakness and hip pain after walking for long distances began at the age of 12. Anti-inflammatory treatment was proposed, which resulted in no improvement, and systemic inflammation was excluded. Physical examination at 17 showed a height of 150 cm (−3.8 s.d.). Multiple joint contracture, short trunk, thoracic kyphosis and swollen interphalangeal joints were noted (Supplementary Figure S1b–d). Laboratory examinations were within the normal range.
Radiological findings indicated decreased heights of the vertebrae (Figure 1f), lessened joint spaces in the hips with heavy cartilage loss, large femoral epiphyses and enlargement of the metaphysis of the interphalangeal joints (Figure 1g–i).
In retrospect, the radiographic findings of the two patients were quite consistent with previous reports of PPD;9–11 however, because of the closure of the growth plates and secondary changes related to aging, a definitive diagnosis was difficult. Therefore, we performed molecular analysis of the patients. We extracted genomic DNA from the peripheral blood using a QIAGEN DNA minikit (Qiagen, Hilden, Germany).
Due to the difficulty in diagnosis, we conducted whole-exome sequencing in Patient 1. In brief, genomic DNA was divided into smaller fragments of 200–250 bp with an ultrasonic instrument (Covaris LE220, Woburn, MA, USA). Ampure Beads (Beckman Coulter, Brea, CA, USA) purification was used to add poly A through a joint reaction at the end of the purified DNA fragments. Agene-trapping chip (Roche NimbleGen, Madison, WI, USA) was used to hybridize and capture the DNA fragments. After hybridization, the captured DNA was sequenced on Illumina HiSeq2500 Analyzers (Illumina, SanDiego, CA, USA) and read on quality analysis pipeline (PIQA).12 We used BWA v0.5913 to align the sequence reads to the human genome reference (build 37) and removed duplicated reads from the subsequent analyses. Sequences variants were identified by comparison with the NCBI reference sequence (NM 198239.1) and annotated by ANNOVAR (http://www.openbioinformatics.org/annovar). The 20× coverage for the RefSeq coding region was 98.13% (Supplementary Figure S2).
Because we noticed the same symptoms in patient 1, we examined patient 2 by direct sequencing for WISP3 instead of whole-exome sequencing. The entire coding region of WISP3 was sequenced using five pairs of primers (the sequences are available on request). Sanger sequencing was then performed in the proband’s family members.
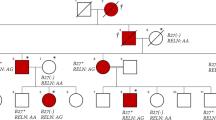
Patient 1 had two novel missense mutations, NM_198239.1 (WISP3_v001):c.396T>G [p.(Cys132Trp)] and NM_198239.1 (WISP3_v001): c.721T>G [p.(Cys241Gly)] (Figure 2a). Both mutations had not been previously reported and were not found in sequencing data projects (Exome Aggregation Consortium Exome; http://exac.broadinstitute.org/). Patient 2 had ahomozygous frameshift mutation, NM_198239.1 (WISP3_v001): c.679dup [p.(Cys227Leufs*21)] (Figure 2b), which has previously been described in two PPD individuals.14 One adenine-ribonucleotide duplication altered the reading frame downstream of codon 227 and caused a premature stop signal (base sequence T-G-A) at codon 247(C227fs*21). SIFT and Polyphen-215 both predicted these mutations to cause severe damage to the WISP3 protein function.

WISP3 mutations in the patients with progressive pseudorheumatoid dysplasia. (a) Family of patient 1. Patient 1 was a compound heterozygote of the mutationsc.396T>G [p.(C132W)]and c.721T>G [p.(C241G)]. Mother of patient 1 was confirmed to have one mutation (c.396T>G of WISP), whereas father and brother of patient 1 were confirmed to have the other mutation (c.721T>G of WISP). This suggested that the two mutations in patient 1 were compound. Neither of the two mutations was present in the 50 controls. (b) Family of patient 2. Patient 2 was a homozygote of the mutation c.679dup [p.(C227Lfs*21)]. The frameshift mutation of the carrier parents needs to be read in theelectropherogram on both the forward and reverse sides. (c) Amino acid alignments in different species around the missense mutation. p.C132and p.C241 are highly evolutionarily conserved.
We performed Sanger sequencing for the two families and confirmed the mutations in the patients and their siblings (Figure 2a,b). In patient 1, the maternal mutation c.396T>G [p.(C132W)] and the paternal mutation c.721T>G [p.(C241G)] were highly conserved among diverse species (Figure 2c).
Thus, we have found two novel WISP3 mutations in cases of PPD. Our study performed the whole-exome sequencing, which allowed us to obtain molecular results faster, especially when traditional specific diagnosis takes a longer time due to sequencing a large gene. The exome allows for the possibility of analyzing the first candidate gene and checking the presence of mutations in other genes at the same time.16,17 Our study has further expanded the WISP3 mutation spectrum and thus contributed to the earlier detection of this rare disease, providing significant benefits to patients’ families and greater awareness of the growing amount of PPD patients for future clinical diagnosis.
References
References
Kahn MF, Corvol MT, Jurmand SH, Couture B, Amouroux J, de Sèze S . Chondrodysplasic rheumatism. Rev Rhum Mal Osteoartic 1970; 37: 825–841.
Garcia Segarra N, Mittaz L, Campos-Xavier AB, Bartels CF, Tuysuz B, Alanay Y et al. The diagnostic challenge of progressive pseudorheumatoid dysplasia (PPRD): a review of clinical features, radiographic features, and WISP3 mutations in 63 affected individuals. Am J Med Genet 2012; 160C: 217–229.
Suwairi WM, Warman ML . Progressive pseudorheumatoid dysplasia. Eur Radiol 2000; 10: 1832–1835.
Bork P . The modular architecture of a new family of growth regulators relatedtoconnective tissue growth factor. FEBS Lett 1993; 327: 125–130.
Hurvitz JR, Suwairi WM, Van Hul W, El-Shanti H, Superti-Furga A, Roudier J et al. Mutationsin the CCN gene family memberWISP3 cause progressive pseudorheumatoiddysplasia. Nat Genet 1999; 23: 94–98.
Rachfal AW, Brigstock DR . Structural and functional properties of CCN proteins. Vitam Horm 2005; 70: 69–103.
Liu L, Li N, Zhao Z, Li W, Xia WB . Novel WISP3 mutations causing spondyloepiphyseal dysplasia tarda with progressive arthropathy in two unrelated Chinese families. Joint Bone Spine 2015; 82: 125–128.
Davis L, Chen Y, Sen M . WISP3 functions as a ligand and promotes superoxide dismutase activity. Biochem Biophys Res Commun 2006; 342: 259–265.
Luo H, Shi C, Mao C . A novel compound WISP3 mutation in a Chinese family with progressivepseudorheumatoid dysplasia. Gene 2015; 564: 35–38.
Wynne-Davies R, Hall C, Ansell BM . Spondyloepiphysealdysplasia tarda with progressive arthropathy. A ‘new’ disorder of autosomal recessive inheritance. J Bone Joint Surg Br 1982; 64: 442–445.
Yue H, Zhang ZL, He JW . Identification of novel mutations in WISP3 gene in two unrelated Chinese families with progressive pseudorheumatoid dysplasia. Bone 2009; 44: 547–554.
Martínez-Alcántara A, Ballesteros E, Feng C, Rojas M, Koshinsky H, Fofanov VY et al. PIQA: pipeline for Illumina G1 genome analyzer data quality assessment. Bioinformatics 2009; 25: 2438–2439.
Li H, Durbin R . Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2010; 26: 589–595.
Ye J, Zhang HW, Qiu WJ, Han LS, Zhang YF, Gond ZW et al. Patients with progressive pseudorheumatoid dysplasia: from clinical diagnosis to molecular studies. Mol Med Rep 2012; 5: 190–195.
Kelly LA, Stemberg MJE . Protein structure on the web: a case study using the Phyre server. Nat Protoc 2009; 4: 363–371.
Lim BC, Yoo SK, Lee S, Shin JY, Hwang H, Chae JH et al. Hoyeraal-Hreidarsson syndrome with a DKC1 mutation identified by whole-exome sequencing. Gene 2014; 546: 425–429.
Mansouri M, Kayserili H, Elalaoui SC, Nishimura G, Iida A, Lyahyai J et al. Novel DDR2 mutation identified by whole exome sequencing in a Moroccan patientwith spondylo-meta-epiphyseal dysplasia,short limb-abnormal calcification type. Am J Med Genet A 2016; 170A: 460–465.
Data Citations
Jiang, Qing HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.903
Jiang, Qing HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.906
Jiang, Qing HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.909
Acknowledgements
This work was supported by the National Science Foundation for Distinguished Young Scholars of China (81125013) and the National Natural Science Foundation of China (81420108021). We thank the patients and families who donated blood samples for this study.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yan, W., Dai, J., Xu, Z. et al. Novel WISP3 mutations causing progressive pseudorheumatoid dysplasia in two Chinese families. Hum Genome Var 3, 16041 (2016). https://doi.org/10.1038/hgv.2016.41
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.41
This article is cited by
-
Unique mutation spectrum of progressive pseudorheumatoid dysplasia in the Chinese population: a retrospective genotype–phenotype analysis of 105 patients
World Journal of Pediatrics (2023)
-
Progressive pseudorheumatoid dysplasia: a rare childhood disease
Rheumatology International (2019)
-
Progressive pseudorheumatoid dysplasia with new-found gene mutation of Wntl inducible signaling pathway protein 3
Pediatric Rheumatology (2018)
-
Delayed-onset of progressive pseudorheumatoid dysplasia in a Chinese adult with a novel compound WISP3 mutation: a case report
BMC Medical Genetics (2017)
