
Abstract
Published genome-wide association studies (GWASs) identified an intergenic region with regulatory features on chr11p13 associated with cystic fibrosis (CF) lung disease severity. Targeted resequencing in n=377, followed by imputation to n=6,365 CF subjects, was used to identify unrecognized genetic variants (including indels and microsatellite repeats) associated with phenotype. Highly significant associations were in strong linkage disequilibrium and were seen only in Phe508del homozygous CF subjects, indicating a CFTR genotype-specific mechanism.
Similar content being viewed by others

Lung disease in cystic fibrosis (CF) varies even among patients homozygous for the same genetic mutation (Phe508del), accounting for ~70% of CF alleles in Caucasian populations.1 Using genome-wide association studies (GWASs) of 6,365 subjects, the International Cystic Fibrosis Gene Modifier Consortium (abbreviated Consortium) has identified genetic loci associated with CF lung disease severity, including an intergenic region on chr11p13.2,3 The region is flanked by genes of high potential relevance to CF lung disease, including: EHF, a transcription factor involved in epithelial differentiation;4,5 and APIP, a dual-function protein with roles in apoptosis and methionine salvage.6,7 The gene PDHX is also in the region and shares a promoter with APIP. Interestingly, the association signal in this region was stronger in 4,139 subjects homozygous for the most common CF mutation (Phe508del) compared with an analysis that included CF individuals with any severe CFTR genotype.2
Targeted resequencing is a viable approach to obtain a detailed genetic variant map to facilitate post-GWAS mechanistic studies. Toward this end, targeted resequencing (termed “ReSeqChr11”) between the 5′ end of EHF and the 3′ end of PDHX (human reference genome assembly (hg19), chr11:34,641,749–35,017,674) was conducted using National Heart, Lung, and Blood Institute (NHLBI) Resequencing and Genotyping Services (rsng.nhlbi.nih.gov) (Figures 1a–c and Supplementary Figure 1). Resequencing was conducted in 377 homozygous Phe508del subjects selected from the larger Genetic Modifier Study cohort (University of North Carolina at Chapel Hill/Case Western Reserve University) that was recruited based on extremes of lung disease severity.8 Samples selected were balanced for gender, lung disease severity (KNoRMA; equal numbers >0.4 and <0.4),9 and by the genotypes of the first reported3 single-nucleotide polymorphism (SNP) with lowest P value, rs12793173, that was found to be in strong linkage disequilibrium (LD) with the GWAS1+22 top SNP, rs10742326 (Supplementary Table 1 and Supplementary Figures 1 and 2).2,3 All library preparation, enrichment, and sequencing were performed at University of Washington Genome Center at Seattle using a custom NimbleGen SeqCap probe library. Resequencing provided coverage of ~81% of the entire region with an average of 259× coverage in the sequenced regions. Gaps in coverage were because of highly repetitive features not compatible with the NimbleGen capture platform (Figure 1c).

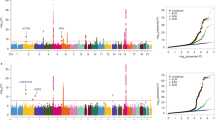
Summary of ReSeqChr11 and cystic fibrosis (CF) lung disease association testing. Annotation information and CF lung disease association test results were converted into either BED, or BEDGRAPH format with hg19 coordinates, and displayed as custom tracks on University of California Santa Cruz (UCSC) genome browser with other relevant public annotations. The sections are: (a) scale bar and genome coordinates on chr11 of UCSC hg19 reference genome; (b) UCSC genes annotation showing EHF, APIP, and PDHX genomic structure; (c) tiled region of NimbleGen probes used to enrich the local genomic DNA to be resequenced; (d) CF lung disease severity association P values for imputed single-nucleotide polymorphisms (SNPs)/indels among Phe508del homozygous patients; (e) CF lung disease severity association P values in non-Phe508del homozygous patients; (f) CF lung disease severity association P values for microsatellite length polymorphisms in Phe508del homozygous patients; (g) Scale bar and genome coordinates on chr11 of UCSC hg19 reference genome for zoom-in region; (h) zoom-in view of genomic locations of SNPs/indels with most significant CF lung disease severity association (P value <10−7), red bar denoted with red asterisk (*) represents the top GWAS1+2 association SNP rs107423262 and blue asterisk (*) two SNPs down denotes top SNP in this study (rs374869483); (i) genomic locations of all SNP/indels in the region with minor allele frequency (MAF) >0.05 colored by linkage disequilibrium (LD) R2 values (R2>0.8, red lines; R2=0.6–0.8, blue lines) compared with rs10742326; (j) DNase I hypersensitivity peak assignments from pHTE cells; (k) DNase I hypersensitivity peak assignments from K562 leukemia cell line; and (l) summary of transcription factor binding motifs from ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) track.
The paired-end 49 bp sequence reads were mapped to reference genome hg19 by BWA v0.5.9-r16.10 SNP and insertion/deletion (indel) calls were made at the University of Washington using an automated software pipeline based on GATK toolkits v1.0-6125.11,12 The initial resequencing variant calls contained 4,800 variants, including SNPs/indels, with National Center for Biotechnology Information (NCBI) dbSNP134 annotation. Results for 94 previously genotyped SNPs were 99.98% concordant. SNP/indel calls were manually reviewed using both quality information from the variant call format (VCF) files and selected spot checks of sequencing read alignment from the binary alignment map (BAM) files, using the Integrative Genomics Viewer (IGV) genome browser.13 When reviewed by manual inspection in Integrative Genomics Viewer, only 40% of indels called by the GATK toolkit were verified as expected.14 The final, manually reviewed, SNP/indel calls from the 377 patient samples contained 2,991 variant calls over the resequenced region. In addition, 101 polymorphic microsatellites were called using GenoTan v0.1.5.15 We also identified a 113-bp deletion, corresponding to rs78669256 (115 bp deletion on hg19; 0.34 minor allele frequency (MAF) in our resequenced samples), that is part of a LINE element whose allele frequency and validation was unknown from single-nucleotide polymorphism database (dbSNP; Supplementary Figure 3).
The 2,991 variants were updated with NCBI dbSNP141 annotation through chromosomal location on hg19 (using ANNOVAR16 and University of California Santa Cruz (UCSC) genome databases), and 946 of them represent novel variants that tended to be rare (Supplementary Figure 4, black dots), with only 1 deletion (rs535729750 in dbSNP142) with MAF >0.1 and only 23 deletions with MAF >0.01.
For association with CF lung disease severity, imputation to the remainder (n=6,365) of the Consortium patient cohort2 was performed using MACH and Minimac2, as previously described,17 except that reviewed variants from ReSeqChr11 were used as the reference set instead of 1000 Genomes variants. For microsatellite repeat polymorphisms, imputation was performed using Beagle v4.018 because of its ability to impute multiple alleles at a single locus. Of the total 3,160 imputed variants over the resequenced region, 1,485 were of sufficient MAF (>0.01) and imputation quality (R2>0.3, Supplementary Figure 4, vertical purple dashed line) to provide reliable tests of association with CF lung disease severity. Association testing was performed as previously described2 with genotype PCs and sex as covariates. Briefly, the Consortium’s quantitative lung phenotype9 was regressed on imputed allele dosages using linear regression for each Consortium cohort,2 followed by a meta-analysis to obtain the final reported random effect P value. Microsatellite repeat variants were coded using the most common allele as the reference, and all other alleles as the alternative alleles before association testing.
Overall, the association P values are highly significant and reflect published GWAS results2,3 (Table 1, Figure 1, and Supplementary Figure 5). The top associated SNPs (P<10−7) for Phe508del homozygous subjects (n=4,139) were located between chr11:34,776,532 and 34,819,022, with the top SNP identified as rs374869483 (Figure 1d and blue asterisk Figure 1h), an indel located 580 bp downstream from the published top SNP (rs10742326)2 (red asterisk, Figure 1h). Importantly, no significant association findings (P<10−5) were identified when the CF cohort was limited to non-Phe508del homozygous subjects (n=2,226) (Table 1 and Figure 1e), and interaction analysis between the top SNP (rs374869483) and Phe508del homozygous status was significant at P=0.046. This is consistent with previous results that reported a reduction in significance of the associations for this region in the entire CF cohort compared with Phe508del homozygous subjects2 (Figure 1d). To evaluate association signals independent of the SNP with lowest P value, the imputed dosage of the conditioning SNP (rs10742326; top SNP in GWAS1+2 that is in LD with rs374869483 (R2=0.987, D′=0.995)) was included in the statistical model as a covariate (Supplementary Figure 6). No independent evidence of association was identified. In addition, SNP-set Kernel Association Test (SKAT) and Burden tests19 applied to the rare variants (MAF <0.01) in EHF, APIP, PDHX, and several regulatory features from the 377 selected samples did not identify any significant associations (Supplementary Table 2). This analysis was limited to these subjects because rare variants cannot be imputed to the entire population.
Together, the analysis suggests that the causal mechanism is driven by one or more common variants in strong LD (Figure 2). Indeed Haploview software (default settings)20 identified strong LD among all highly associated variants (Figures 1g–i and 2a–d). The top 5 haplotypes containing the 111 highly associated SNPs are shown in Figure 2e, with 3 most common haplotypes representing 38, 24, and 22% of all haplotypes. The fact that variants associated with disease are often in high LD and occur in context of local haplotype structure raised the possibility that the mechanism of regulation acts through multiple sites.

Linkage disequilibrium (LD) and haplotype structure around top cystic fibrosis (CF) lung disease-associated region. The upper panels (a–d) show the entire CF lung disease severity association region, whereas the lower panel (e) indicates the LD structure and the top 5 haplotypes of single-nucleotide polymorphisms (SNPs) with the most significant association P value (<10−7). The sections are: (a) scale bar and genome coordinates on chr11 of University of California Santa Cruz (UCSC) hg19 reference genome; (b) CF lung disease severity association P values; (c) LD plot SNP locations with respect to the genome coordinates in (a) (upward tick marks) that are then mapped to the LD plot in (d) (slanted lines); (d) LD plot generated by Haploview; (e) haplotype structure with allele genotypes and frequencies; the first SNP (rs77939918) and last SNP (rs7120959) are labeled in black font with black arrows at the bottom; the SNP of highest significance (rs10742326) is labeled in red font with a red arrow. Asterisks (*) indicate common alleles observed in the top five haplotypes.
The intergenic nature of this region suggests it acts through complex gene regulatory functions. Regulatory features downloaded from SeattleSeq Annotation 138, UCSC genome browser, ENCODE (http://www.genome.gov/10005107),21–25 and Roadmap Epigenomics servers, and annotation information over the resequenced region on chr11, were collated by chromosomal locations on hg19 (Supplementary Table 3).26 Careful examination reveals complex, multiple, cell-type-specific regulatory features; for example, DNase I hypersensitive sites have been documented from human tracheal epithelium27 that are distinct from those identified in K562 cells (Figure 1j vs. k). The two DNase I hypersensitive sites in human tracheal epithelium (11.2524 and 11.2525)27 contain a large number of transcription factor binding sites (including four FOXA1 binding sites), as identified in chromatin immunoprecipitation sequencing assays (Figure 1l and Supplementary Table 4). In addition, epigenetic markers found in Calu3 cell line (goblet cell model from human lung adenocarcinoma) point to potential transcription enhancer activity overlapping the two FOXA1 binding segments (Supplementary Table 3). These features are potentially relevant given the known roles for FOXA1 in mucin production.28,29 Furthermore, the highly significant SNP rs10742325 (Table 1) has been found to show an allele-specific DNase I footprint (false discovery rate <0.05).26
Although our catalog of observed variants is still incomplete because of gaps in sequence coverage and present limitations of alignment and variant calling from short sequence reads, these results suggest that there is no obvious single sequence variant that is driving the association in the region. The mechanism ultimately must explain how the regulatory features operate in the context of cell-specific effects and explain the Phe508del homozygous-specific association observed in this region.
References
References
Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H et al. The relation between genotype and phenotype in cystic fibrosis- analysis of the most common mutation (deltaF508). New Engl J Med 1990; 323: 1517–1522.
Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR et al. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun 2015; 6: 8382.
Wright FA, Strug LJ, Doshi VK, Commander CW, Blackman SM, Sun L et al. Genome-wide association and linkage identify modifier loci of lung disease severity in cystic fibrosis at 11p13 and 20q13.2. Nat Genet 2011; 43: 539–546.
Fossum SL, Mutolo MJ, Yang R, Dang H, O'Neal WK, Knowles MR et al. Ets homologous factor regulates pathways controlling response to injury in airway epithelial cells. Nucleic Acids Res 2014; 42: 13588–13598.
Stephens DN, Klein RH, Salmans ML, Gordon W, Ho H, Andersen B . The Ets transcription factor EHF as a regulator of cornea epithelial cell identity. J Biol Chem 2013; 288: 34304–34324.
Kang W, Hong SH, Lee HM, Kim NY, Lim YC, Le le TM et al. Structural and biochemical basis for the inhibition of cell death by APIP, a methionine salvage enzyme. Proc Natl Acad Sci USA 2014; 111: E54–E61.
Cho DH, Hong YM, Lee HJ, Woo HN, Pyo JO, Mak TW et al. Induced inhibition of ischemic/hypoxic injury by APIP, a novel Apaf-1-interacting protein. J Biol Chem 2004; 279: 39942–39950.
Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F et al. Gene modifiers of lung disease in cystic fibrosis. New Engl J Med 2005; 353: 1443–1453.
Taylor C, Commander CW, Collaco JM, Strug LJ, Li W, Wright FA et al. A novel lung disease phenotype adjusted for mortality attrition for cystic fibrosis genetic modifier studies. Pediatr Pulmonol 2011; 46: 857–869.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011; 43: 491–498.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G et al. Integrative genomics viewer. Nat Biotechnol 2011; 29: 24–26.
Hasan MS, Wu X, Zhang L . Performance evaluation of indel calling tools using real short-read data. Hum Genomics 2015; 9: 20.
Tae H, Kim DY, McCormick J, Settlage RE, Garner HR . Discretized Gaussian mixture for genotyping of microsatellite loci containing homopolymer runs. Bioinformatics 2014; 30: 652–659.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 2007; 316: 1341–1345.
Browning SR, Browning BL . Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 2007; 81: 1084–1097.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X . Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet 2011; 89: 82–93.
Barrett JC, Fry B, Maller J, Daly MJ . Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 2005; 21: 263–265.
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489: 57–74.
Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C et al. Architecture of the human regulatory network derived from ENCODE data. Nature 2012; 489: 91–100.
Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 2012; 22: 1798–1812.
Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res 2013; 41: D171–D176.
Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013; 41: D56–D63.
Maurano MT, Haugen E, Sandstrom R, Vierstra J, Shafer A, Kaul R et al. Large-scale identification of sequence variants influencing human transcription factor occupancy in vivo. Nat Genet 2015; 47: 1393–1401.
Bischof JM, Ott CJ, Leir SH, Gosalia N, Song L, London D et al. A genome-wide analysis of open chromatin in human tracheal epithelial cells reveals novel candidate regulatory elements for lung function. Thorax 2012; 67: 385–391.
Ye DZ, Kaestner KH . Foxa1 and Foxa2 control the differentiation of goblet and enteroendocrine L- and D-cells in mice. Gastroenterology 2009; 137: 2052–2062.
van der Sluis M, Vincent A, Bouma J, Korteland-Van Male A, van Goudoever JB, Renes IB et al. Forkhead box transcription factors Foxa1 and Foxa2 are important regulators of Muc2 mucin expression in intestinal epithelial cells. Biochem Biophys Res Commun 2008; 369: 1108–1113.
Data Citations
Dang, Hong HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.838
Dang, Hong HGV Database (2016) http://dx.doi.org/10.6084/m9.figshare.hgv.841
Acknowledgements
Genotyping services were provided by the Northwest Genomics Center at the University of Washington, Department of Genome Sciences, under US Federal Government contract number HHSN268201100037C from the National Heart, Lung and Blood Institute. We thank Dr Debra Nickerson for her expertise with regard to the original patient population selection, region to be sequenced, and for discussions with regard to indel calls. We thank Dr Ann Harris and Dr Fred Wright for expertise in study design and analysis. In addition, support was provided by the National Heart, Lung and Blood Institute (HL117843, HL11998, and HL068890), the National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK065988), and the Cystic Fibrosis Foundation (KNOWLE00A0 and R026).
Author information
Authors and Affiliations
Contributions
Study design: WKO and MRK; patient recruitment and phenotype collection: HC, GRC, MLD, MRK, RGP, JRS, and LJS; resequencing data collection and analysis: HD, PJG, RGP, XG, JRS, MRK, and WKO; manuscript preparation: HD, RGP, JRS, MRK, and WKO.
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv).
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Dang, H., Gallins, P., Pace, R. et al. Novel variation at chr11p13 associated with cystic fibrosis lung disease severity. Hum Genome Var 3, 16020 (2016). https://doi.org/10.1038/hgv.2016.20
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2016.20
