
Abstract
Cetuximab, a monoclonal antibody against epidermal growth factor receptor (EGFR), was shown to be active in colorectal cancer. Although some patients who harbor K-ras wild-type tumors benefit from cetuximab treatment, 40 to 60% of patients with wild-type K-ras tumors do not respond to cetuximab. Currently, there is no universal marker or method of clinical utility that could guide the treatment of cetuximab in colorectal cancer. Here, we demonstrate a method to predict response to cetuximab in patients with colorectal cancer using OncoFinder pathway activation strength (PAS), based on the transcriptomic data of the tumors. We first evaluated our OncoFinder pathway activation strength model in a set of transcriptomic data obtained from patient-derived xenograft (PDx) models established from colorectal cancer biopsies. Then, the approach and models were validated using a clinical trial data set. PAS could efficiently predict patients’ response to cetuximab, and thus holds promise as a selection criterion for cetuximab treatment in metastatic colorectal cancer.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in the United States. The American Cancer Society estimates that, in 2015, 132 700 people will be diagnosed with CRC and that 49 700 people will die from the disease. Distant metastasis is the main cause of death in CRC patients, and 40–50% of newly diagnosed patients are already in advanced stages when diagnosed.1 In the past decade, the management of patients with metastatic CRC (mCRC) has been profoundly improved by the introduction of anti-epidermal growth factor receptor (anti-EGFR) monoclonal antibodies, cetuximab (Erbitux) and panitumumab (Vectibix). Clinical trials have shown the activity of cetuximab as a single agent and in combination with chemotherapeutic agents in advanced CRC.2–5
It is well established that K-ras mutation status is a strong predictive factor for anti-EGFR therapy in patients with mCRC. Although anti-EGFR therapy has little or no effect in colorectal tumors harboring K-ras mutations (codons 12 and 13 in the exon 2), patients with wild-type K-ras tumors are more likely to benefit from the treatment.6,7 However, K-ras wild-type status is not a reliable predictor of tumor response to anti-EGFR monoclonal antibodies, as only about 40–60% of patients with wild-type K-ras benefit from anti-EGFR therapy.6,7
EGFR orchestrates various processes involved in cell growth, differentiation, survival, cell cycle progression, angiogenesis and drug sensitivity via Ras-Raf-MAPK, PI3K-AKT, JAK/STAT and other pathways.8 Therefore, accumulative evidence suggests that an increase in the EGFR gene copy number and dysregulation of downstream EGFR signaling pathway modulators, such as BRAF, HRAS, NRAS, PI3K and AKT/PTEN, are also important factors when determining tumor sensitivity to EGFR antibodies.9,10 Previous studies have demonstrated that neither EGFR activation nor EGFR expression level itself is capable of discriminating responses to cetuximab in CRC.11–13 Moreover, EGFR mutations are rare in CRC and have no clinical relevance with regard to the activity of anti-EGFR therapy.14,15 Although multiple efforts have been made to identify additional biomarkers to predict cetuximab response in wild-type K-ras CRC,7,16–19 no reliable markers of clinical utility have been identified. Therefore, there is an urgent need to develop new strategies to identify patients whose tumors could respond to and clinically benefit from anti-EGFR therapy in mCRC.
We hypothesized that analysis of the comprehensive tumor pathway activation profile may be a more efficient strategy to segregate cetuximab responders from non-responders in the K-ras wild-type population than previously described methods, such as evaluating the gene expression profile,16 selective pathways expression status19 or genotyping EGFR downstream effectors for activating mutations.18 As a novel approach to improving the decision-making in the treatment of solid cancers, we propose a new in silico drug screening and efficacy prediction tool, OncoFinder, for both quantitative and qualitative analysis of the intracellular signaling pathway activation.20,21 OncoFinder performs pathway-level analysis of an expression data set of tumors and determines the pathway activation strength (PAS). PAS is a measurement of the cumulative value of perturbations of a signaling pathway and serves as a valuable cancer biomarker.20–22
In the current study, this approach was extensively evaluated for the prediction of cetuximab sensitivity using the expression microarray data set from patient-derived CRC tumorgrafts and validated in a cohort of CRC patient data available from a Phase II exploratory clinical trial. TumorGrafts or patient-derived xenografts are established from directly implanted tumor tissue samples into an immunodeficient mouse. TumorGrafts are increasingly recognized as representative in vivo clinical models and are vastly superior to commonly used cell line xenografts.23–26 TumorGraft or patient-derived xenograft models maintain global gene expression patterns, DNA copy-number alterations, mutational status, metastatic potential, clinical predictability and tumor architecture of the parental primary tumors.25,27 Therefore, personalized tumorgrafts can be successfully used as model platforms for drug screening and improving decision-making in tumor treatment. Time is critical for definitive treatment, especially for advanced cancer patients, and the entire process of implantation and propagation followed by drug screening typically takes 12–16 weeks. As OncoFinder could increase the therapy success and decrease the time and cost for effective tumorgraft drug screening by narrowing down the drug candidates, we first evaluated whether the OncoFinder PAS algorithm can predict cetuximab sensitivity in a set of transcriptomic data obtained from CRC tumorgrafts and then validated our approach in CRC patient data available from a clinical trial. Taken together, our study demonstrates that PAS was capable of predicting the cetuximab-sensitive tumor phenotype in both tumorgrafts and primary human tumors. Furthermore, the combined predictive value of PAS and K-ras mutation status could predict the cetuximab response more accurately than either PAS or K-ras as stand-alone markers. These observations have important clinical implications for the treatment of patients with EGFR inhibitors, as PAS may have clinical value as a predictive biomarker to discern patients who are likely to benefit from EGFR inhibitors from those who are unlikely to respond to such therapy.
Materials and Methods
Gene expression and drug response of tumorgrafts and human CRC
Before cetuximab treatment, the gene expression of 92 CRC tumorgrafts derived from 33 patients was investigated using microarray. Raw data (CEL files) and tumor growth inhibition (TGI) data from six patients were obtained through collaboration with Champions Oncology using their extensive internal gene expression database. To avoid any platform-dependent variation, as a reference we used 10 mucosa samples from healthy donors obtained from the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo) repository data set GSE44076 (sample GSM1077598-GSM1077607) produced on the same platform.28 Human CRC gene expression data sets containing both healthy colorectal samples and tumor samples were selected. Three cohorts of colorectal patient samples were downloaded from GEO (GEO accession: GSE21510, GSE33113, and GSE44076). PAS values were calculated for each pathway and each sample in both tumorgrafts and cohorts of human CRC patients. Then, the PAS values of the tumorgrafts were compared with the PAS values of each cohort of human CRC samples. Correlations were computed between every two sets of PAS values. Finally, the linear regressions were applied to the correlations. As a validation data set, we used a phase II exploratory pharmacogenomics study containing eighty patients (n=80) with mCRC treated with cetuximab (GEO accession: GSE5851).17
Bioinformatics analysis and expression data pre-processing
All microarray preprocessing steps were performed in R version 3.1.0 using packages from Bioconductor.29 Raw microarray data (CEL files) from tumors and samples from healthy donors were pre-processed with the GCRMA algorithm using the affy package30 and summarized using redefined probe set definition files from the Brainarray repository (Version 17).31 Obtained gene expression values were averaged across all replicates.
OncoFinder PAS
Preprocessed gene expression data were loaded into OncoFinder software suite. PAS serves to evaluate the degree of pathological changes in the signaling pathway. The algorithm used to calculate PAS is as follows:
Here, CNRn is the ratio of the expression level of a gene n in the tumor sample and in the control; BTIFn is a value of beyond tolerance interval flag, which equals 0 or 1; and ARRn is an activator/repressor role equal to −1, −0.5, 0, 0.5 or 1, defined by the role of protein n in the pathway. More information can be found in previous publications.20,21 PASs were determined using the default parameters of OncoFinder, a sigma filter of 2 and a CNR value <0.67 or >1.5.
Principal component analysis
Principal component analyses were performed to examine any variation and clustering between PAS of tumorgrafts and GSE44076 using the prcomp function of the ‘stats’ package in R.
Linear prediction model training in CRC tumorgrafts
PASs were prepared as outlined above. A linear regression model was fitted for tumorgrafts TGI against PAS. An R package ggplot2 from Bioconductor was used to generate the linear equations and plot the graphs.
The area under the ROC curve
The area under the ROC curve values were calculated according to Brisov et al.22 and Subramanian and Simon.32 Statistical analyses were performed using the R package.
Validation of the model in a CRC clinical trial
For the CRC clinical trial, all gene expression data were preprocessed and PASs were determined using OncoFinder, as described above. First, the tumorgraft-trained linear models were used to calculate a predicated TGI value for each patient. Then, the predicated TGI values were compared with the patients’ progression-free survival (PFS) values. A Pearson’s correlation test was used to estimate the accuracy and significance of the prediction.
Results
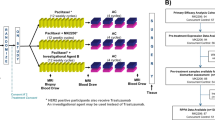
This multistage study was designed to investigate a novel approach to predicting patients’ response to cetuximab in mCRC. A workflow of the study design is shown in Figure 1. Detailed information about the study design and analytical approach can be found in the Materials and Methods section.

Workflow diagram of the study design and analytical approach for predicting patients’ drug sensitivity. The raw microarray gene expression data were (1) preprocessed using R packages. Then, the PAS for each sample was (2) determined using OncoFinder with the default parameters. (3) A linear regression model was fitted for TGI against the PAS, and then this model was applied to human clinical samples to estimate the predicted TGI. The predicted TGIs for patients were plotted with PFS to determine the accuracy and significance in the prediction of patients’ drug sensitivity (4). More details are available in the Materials and Methods section.
TumorGrafts retain PAS profiles inherent to human CRC
To evaluate the pathway activation profiles of CRC, we first analyzed and compared the pathway activation profiles of tumorgrafts and primary colorectal tumors. Ninety-two tumorgraft samples from 33 independent models were profiled on the Affymetrix Human Genome U219 array platform before treatment with cetuximab. As parental tumor samples were not available for comparison with the tumorgrafts, we chose three cohorts of CRC patient samples from NCBI GEO, GSE21510 with 123 patients, GSE33113 with 90 patients and GSE44076 with 98 patients. None of the patients had been treated with chemotherapy or radiation before their tumor biopsy, so the spectrum of differentially expressed genes observed in these samples largely reflects tumors in their naturally occurring state. The expression microarrays of tumorgrafts and human CRC samples were first normalized and preprocessed with the GCRMA algorithm using R packages. Then, using OncoFinder we determined a quantitative measure of the signaling PAS for the 273 distinct signaling pathways implicated in cancer.22,33 Our comprehensive analysis revealed that 194, 233, 145 and 213 pathways were significantly dysregulated (P value<0.05) in tumorgrafts and each of the three primary human cancer cohorts (GSE21510, GSE33113 and GSE44076 respectively), when these samples were compared with healthy human colonic samples. Overall, we identified 84 distinct signaling pathways commonly dysregulated in all four data sets (Supplementary Table S1 and Supplementary Figure F1). Interestingly, a subsequent analysis of commonly dysregulated signaling pathways revealed an upregulation of the pathways that were shown to be frequently activated in CRC, such as AKT/mTOR, MAPK, RAS, p53 and Wnt.34–37 Moreover, pathway activation profiles of these 84 dysregulated pathways significantly correlated between the tumorgraft models and each one of the primary human colorectal patient cohorts, GSE21510, GSE33113 and GSE44076. The correlation coefficients for tumorgrafts and the GSE21510, GSE33113 or GSE44076 cohorts were 0.7098, 0.5589 and 0.5543, respectively, and all of the correlations had a P-value lower than 0.0001 (Figures 2a–c).

Correlation of pathway activation profiles in CRC tumorgrafts and primary colorectal tumors and principal components analysis. Compared with normal colorectal tissue, significantly upregulated or downregulated pathways were identified based on PAS values (P value<0.05). The PAS values of tumorgrafts correlated significantly with the PAS values of primary colorectal cancer patients in all three cohorts tested: (GSE33113, a), (GSE21510, b) and (GSE44076, c). Principle component analyses (PCA) were performed to assess the variation and clustering between PAS of tumorgrafts and primary CRC patients (GSE44076), and the first three principal components are shown (d). Each sample is represented by one dot. Samples from tumorgrafts (red dots) and the primary CRC data set (GSE44076) (green dots) are overlaid. One set of lung cancer (GSE30219, blue dots) and melanoma (GSE7533, orange dots) samples were also plotted as references. The mean Euclidean distances between the colon cancer tumorgrafts group and the human colon cancer, lung cancer and melanoma groups are 41.43, 79.95 and 124.65, respectively.
To further compare the pathway activation profiles between tumorgrafts and human CRC, principle component analyses were performed to assess any variation and clustering between the PAS of tumorgrafts and primary CRC using the prcomp function of ‘stats’ package in R. Gene expression profiles of patients in cohort GSE44076 were used as representatives of human CRC. As references, pathway activation profiles were calculated from two microarray expression data sets derived from patients with lung cancer (GSE30219) and melanoma (GSE7533) and compared with the results discovered in colorectal tumorgraft models. The score plots were used to assess the clustering between the colorectal tumorgrafts and human CRC, lung cancer or melanoma samples (Figure 2d). The mean Euclidean distances between the colon cancer tumorgrafts group and human colon cancer, lung cancer and melanoma cohorts were 41.43, 79.95 and 124.65, respectively. The first three principal component plotters showed that tumorgrafts were close to and overlaid with human colorectal samples, whereas lung cancer and melanoma samples, which were plotted as references, showed no clustering with either colorectal tumorgrafts or primary colorectal tumors (Figure 2d). These data suggest that pathway activation profiles of the tumorgrafts and primary human CRC can be attributed to collection from divergent random mating populations.
We next compared the PAS values of four representative pathways that are highly associated with EGFR signaling (EGFR1, RAS, MAPK and p53 pathways) between the tumorgrafts and GSE44076 cohort (Figure 3). Despite the relatively small number of tumorgrafts models available for this study (33 CRC tumorgrafts), our analysis determined that the PAS values of the four pathways compared were within a very similar range. Collectively, these results demonstrate that PAS profiles generated from tumorgrafts are highly representative of PAS profiles in primary human CRC at both global and local levels.

Correlation between the PAS values of representative pathways in tumorgrafts and primary colorectal cancers. PASs for the EGFR1 pathway (a), RAS pathway (b), MAPK signaling pathway (c) and p53 signaling pathway (d) were compared in colorectal cancer tumorgrafts and the human colorectal cancer cohort (GSE44076).
Pathway activation profile correlates with cetuximab-sensitivity in colorectal tumorgrafts models
We next used six of the 33 tumorgrafts models, which were treated with cetuximab and for which TGI values were available, to investigate whether the PAS values obtained from analysis of the tumorgrafts could be used to predict cetuximab response. TGI values were calculated following standard procedures.24,25 Two hundred and seventy-three PASs were assessed using Pearson correlations against the TGI values of the tumorgrafts. Our analysis discovered that the PAS of 26 pathways significantly correlated with cetuximab-induced TGI values (P value <0.05) (Supplementary Table S2). Two of the pathways highly associated with CRC carcinogenesis, IL1038–40 and the VEGF-mTOR41–45 pathways, were selected for further analysis, and their PAS values were plotted against the TGIs. Linear regressions were applied to the grafts (regression model: y=16.76*x−0.5848 and y=63.05*x−61.13, respectively) (Figure 4). The PAS of the two selected pathways had a significant positive correlation to the TGI of the tumorgrafts (R2=0.8754, P value=0.0061 and R2=0.7166, P value=0.0335, respectively). Thus, our data indicate that cetuximab-induced TGI in CRC tumorgrafts could be predicted from the PAS of the same tumorgraft models.

Correlation of PAS and tumor growth inhibition (TGI) in colorectal cancer tumorgrafts. Cetuximab-induced TGI in six colorectal cancer tumorgraft models significantly correlated with PAS values of the IL10 pathway (a) and the VEGF-mTOR pathway (b).
Cetuximab treatment in CRC patients
Finally, to validate our approach, we identified linear PAS-TGI models for patients from an available clinical trial data set, which assessed the response to cetuximab monotherapy in 80 patients (n=80) with mCRC (GEO accession: GSE5851).17 In the original study, it was found that patients without K-ras mutations whose tumors expressed high transcriptional levels of the EGFR ligands epiregulin and amphiregulin were more likely to respond to cetuximab.17 As low expression of epiregulin and amphiregulin does not necessarily correlate with EGFR pathway deactivation, which can be upregulated due to activating mutations in downstream pathway targets, we thought that a comprehensive analysis of all cancer-related pathways in the tumor might be a more reliable predictive biomarker of the response to EGFR TKIs.
Interestingly, while the cetuximab-induced TGI, predicted from the PAS values of IL10 and VEGF-mTOR pathways generated from tumorgrafts, failed to correlate with PFS in all treated patients (Figure 5a) and in the K-ras mutant population (Figure 5b) (P values 0.2132, 0.5020 and 0.1403, and 0.8931, respectively), the predicted TGI significantly correlated with PFS in the K-ras wild-type patients (P values 0.0243 and 0.0426, respectively, regression models: y=0.2506*x+93.91 and y=0.4760*x−17.45, respectively) (Figure 5c). Although our data clearly support the fact that K-ras status is a critical factor in predicting cetuximab sensitivity in CRC, it also suggest that our OncoFinder prediction tool may further stratify the patients who probably will not respond to cetuximab from a larger number of K-ras wild-type patients who will respond to cetuximab treatment.

PAS generated in colorectal tumorgrafts can predict cetuximab response in K-ras wild-type CRC patients. Months of progression-free survival were plotted against the TGI values predicted from the PAS of the IL10 pathway (left) and the VEGF-mTOR pathway (right) in all patients (a), K-ras mutant patients (b) and K-ras wild-type (WT) patients (c).
To further assess PAS as a predictive biomarker for cetuximab sensitivity in CRC, the PASs of 273 cancer-related pathways were assessed using Pearson correlations against the patients’ PFS in all 80 patients in this cohort. Our analysis revealed that the PAS values of 18 distinct pathways significantly correlated with PFS (Supplementary Tables S3 and S4). Interestingly, of these 18 pathways, signaling pathways associated with apoptosis negatively correlated with PFS values (Supplementary Table S3), further supporting the credibility of our approach.
To compare PAS and K-ras status as a drug response prediction biomarker for cetuximab in CRC, patients were classified as responders or non-responders. Patients with complete response, stable disease and partial response were defined as responders, whereas patients with progressive disease were defined as non-responders. The PAS values of the two pathways that most significantly correlated with PFS, the JNK pathway (insulin signaling) and the mitochondrial apoptosis pathway (apoptosis), were plotted against cetuximab response (Figures 6a and b). Moreover, the K-ras status of the tumor was plotted against the PFS values of the same patient’s cohort (Figure 6c). As expected, the patients’ K-ras status was significantly correlated with drug response (PFS). Interestingly, although the PASs of both representative pathways were able to significantly discriminate cetuximab responders from non-responsive patients (Figures 6a and b), the ability of both PAS values to predict cetuximab sensitivity was comparable or even better than the predictive value of the K-ras status (Figure 6c). To further evaluate the prognostic power of individual PAS to predict cituximab responsiveness, we performed area under the ROC curve analysis for the PAS values of the 18 distinct pathways that significantly correlated with PFS (Supplementary Table S5). Consistent with previous results, the JNK pathway (insulin signaling) and mitochondrial apoptosis pathway (apoptosis) had area under the ROC curve values of 0.79 and 0.70, respectively. Collectively, our data indicate that individual PAS values are strongly associated with PFS and may represent a prognostic signature for cetuximab responsiveness in CRC patients.

PAS serves as a robust independent predictive biomarker for cetuximab response in colorectal cancer patients. PAS values of the two pathways most significantly correlated with PFS, the JNK pathway (insulin signaling) (a) and the mitochondrial apoptosis pathway (apoptosis) (b) or K-ras status (c) were plotted against cetuximab response for all patients in the cohort. PAS values of the JNK pathway (insulin signaling) (d) or the VEGF-mTOR pathway (e) were plotted against the patients’ response to cetuximab in the K-ras wild-type population.
We next asked whether PAS could further distinguish cetuximab-resistant patients with wild-type K-ras. Our analysis indicates that PAS values of the JNK pathway (insulin signaling) (Figure 6d) and the mitochondrial apoptosis pathway (apoptosis) (Figure 6e) significantly correlated with response to cetuximab in K-ras wild-type CRC patients. Consequently, our data suggest that the concurrent evaluation of K-ras mutation status and PAS may better predict response to cetuximab than either of the factors as stand-alone biomarkers.
Discussion
The treatment of mCRC has evolved significantly over the past decade, and overall patient survival has nearly tripled. A significant contribution to the improvement was the development of novel targeted agents, such as the anti-EGFR monoclonal antibodies cetuximab and panitumumab. Although EGFR is known to be overexpressed in various tumors of epithelial origin, including CRC, multiple independent studies have shown that EGFR activation and expression levels were not capable of predicting cetuximab response in colorectal patients.5,11–13 Furthermore, activating mutations in EGFR are uncommon in CRC and have no clinical relevance for anti-EGFR therapy.14,15
Activating K-ras mutations have been established to be strong predictive biomarkers of cetuximab response, hence the FDA approved the use of cetuximab for the treatment of K-ras mutation-negative (wild-type), EGFR-expressing mCRC. Although the assessment of K-ras mutation status has been included in clinical guidelines for patients with CRC, a significant fraction of patients with wild-type K-ras fail to respond to cetuximab (http://www.cancer.gov/cancertopics/druginfo/fda-cetuximab). Therefore, multiple efforts have been made to detect additional biomarkers to stratify the subset of patients whose tumors could respond and who would clinically benefit from cetuximab therapy from patients whose tumors would not respond. Khambata and Ford suggested that high transcriptional levels of the EGFR ligands epiregulin and amphiregulin might serve as an indicator of cetuximab sensitivity in wild-type K-ras CRC patients.17 However, low expression of epiregulin and amphiregulin is not a reliable indicator of EGFR pathway deactivation, which can be upregulated by activating mutations in downstream pathway targets.
As RAS/RAF/MEK/MAPK and PIK3CA/PTEN/AKT are the key downstream components of the EGFR signaling pathway, several studies have focused on the downstream effectors of EGFR signaling, including other RAS family members, BRAF, PIK3CA and PTEN.9,10,18,19,46–49 In theory, overexpression of the downstream effectors of the EGFR signaling pathway or activating mutations in these genes would likely make anti-EGFR therapy ineffective. While these studies define certain molecular features of cetuximab response, their use as a modality for large-scale implementation has recognized limitations. Multifaceted EGFR signaling affects numerous cellular processes, such as growth, differentiation, survival, cell cycle progression, angiogenesis and drug sensitivity.8 Moreover, extensive cross-talk and transactivation have been observed between EGFR and other RTKs that modulate progression of solid cancers.45,50–57 Thus, assessing the expression status of selective pathways may reveal only a minority of dysregulated signaling processes that are present in the tumor and may therefore underestimate responses to therapy. As an alternative to evaluating selective pathways or genotyping EGFR downstream effectors for activating mutations, the series of independent studies proposed microRNAs as a potential biomarker to predict cetuximab response in CRC patients. It was found that several microRNAs were associated with CRC development, progression and clinical response,58–61 whereas members of the Let-7 microRNA family were reported to be associated with cetuximab sensitivity.62 Although these data look promising, larger confirmatory studies are warranted to further investigate the correlation between microRNAs and drug response in CRC.
Balko and Black suggested that genome-wide gene expression analysis make better markers of cancer than expression of individual genes. Using the transcriptomic data from 100 CRC patients treated with cetuximab, published by Khambata-Ford and colleagues,17 the authors proposed a gene expression model capable of predicting cetuximab response in the K-ras wild-type population.16 Despite its potential clinical utility, the main limitation of this algorithm is the analysis of gene expression without considering the functional roles of the genes in well-defined signaling pathways.
We hypothesized that annotating differentially expressed genes into functional pathways would allow a comprehensive analysis of signaling pathway activation profiles and would offer better predictive capacity and broader clinical utility than raw gene expression evaluation. We have recently developed OncoFinder, a novel tool for quantitative and qualitative analysis of the intracellular signaling pathway activation.20,21 OncoFinder performs pathway level analysis of an expression data set and determines PAS, a measurement of the cumulative value of perturbations of a signaling pathway, which serves as a valuable cancer biomarker.20–22 We have shown here that our approach can predict cetuximab sensitivity in a set of transcriptomic data obtained from CRC tumorgrafts and can be applied to forecast the clinical outcome in patients with mCRC. Successful application of our model to predict the clinical outcome for cetuximab in a cohort of mCRC patients using a PAS profile derived from microarray gene expression data represents a true validation of the predictive strength of the OncoFinder PAS algorithm. Surprisingly, the ability of PAS to predict cetuximab sensitivity in cancer patients was as good as or even better than the predictive value of the K-ras mutation status. Although these data suggest that the PAS is a reliable predictive biomarker of the primary tumor response to cetuximab regardless of K-ras status, the concurrent evaluation of K-ras mutation status and PAS may more accurately predict response to cetuximab than either PAS or K-ras as stand-alone biomarkers. Moreover, OncoFinder PAS was useful for stratifying cetuximab response in K-ras wild-type patients. These data have important clinical implications, since to date, there are no reliable clinical biomarkers to identify wild-type K-ras CRC patients who will benefit from cetuximab therapy.
Although additional studies are warranted to validate the predictive capacity of our model in larger cohorts, our data identify the OncoFinder PAS as a promising predictive biomarker of the response of colorectal tumors to cetuximab and suggest that it should be used in combination with K-ras status.
References
Cancer Facts and Statistics. More information is available online: http://www.cancer.org/research/cancerfactsstatistics/. The American Cancer Society 2014.
Sobrero AF, Maurel J, Fehrenbacher L, Scheithauer W, Abubakr YA, Lutz MP et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 2311–2319.
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 2007; 25: 1658–1664.
Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med 2007; 357: 2040–2048.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–345.
Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26: 374–379.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757–1765.
Kandasamy K, Mohan SS, Raju R, Keerthikumar S, Kumar GS, Venugopal AK et al. NetPath: a public resource of curated signal transduction pathways. Genome Biol 2010; 11: R3.
De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S . KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 2011; 12: 594–603.
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 2009; 27: 5924–5930.
Shia J, Klimstra DS, Li AR, Qin J, Saltz L, Teruya-Feldstein J et al. Epidermal growth factor receptor expression and gene amplification in colorectal carcinoma: an immunohistochemical and chromogenic in situ hybridization study. Modern Pathol 2005; 18: 1350–1356.
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 2005; 23: 1803–1810.
Saltz LB, Meropol NJ, Loehrer PJ Sr, Needle MN, Kopit J, Mayer RJ . Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol 2004; 22: 1201–1208.
Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA . Responsiveness to cetuximab without mutations in EGFR. N Engl J Med 2005; 353: 208–209.
Mendelsohn J, Baselga J . Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 2003; 21: 2787–2799.
Balko JM, Black EP . A gene expression predictor of response to EGFR-targeted therapy stratifies progression-free survival to cetuximab in KRAS wild-type metastatic colorectal cancer. BMC cancer 2009; 9: 145.
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 2007; 25: 3230–3237.
Pentheroudakis G, Kotoula V, De Roock W, Kouvatseas G, Papakostas P, Makatsoris T et al. Biomarkers of benefit from cetuximab-based therapy in metastatic colorectal cancer: interaction of EGFR ligand expression with RAS/RAF, PIK3CA genotypes. BMC cancer 2013; 13: 49.
Jhawer M, Goel S, Wilson AJ, Montagna C, Ling YH, Byun DS et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 2008; 68: 1953–1961.
Buzdin AA, Zhavoronkov AA, Korzinkin MB, Venkova LS, Zenin AA, Smirnov PY et al. Oncofinder, a new method for the analysis of intracellular signaling pathway activation using transcriptomic data. Front Genet 2014; 5: 55.
Buzdin AA, Zhavoronkov A, Korzinkin M, Roumiantsev SA, Aliper AM, Venkova LS et al. The OncoFinder algorithm for minimizing the errors introduced by the high-throughput methods of transcriptome analysis. Front Mol Biosci 2014; 1.
Borisov NM, Terekhanova NV, Aliper AM, Venkova LS, Smirnov PY, Roumiantsev S et al. Signaling pathway activation profiles make better markers of cancer than expression of individual genes. Oncotarget 2014; 5: 10198–10205.
Garber K . Personal mouse colonies give hope for pancreatic cancer patients. J Natl Cancer Inst 2007; 99: 105–107.
Morelli MP, Calvo E, Ordonez E, Wick MJ, Viqueira BR, Lopez-Casas PP et al. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J Clin Oncol 2012; 30: e45–e48.
Hidalgo M, Bruckheimer E, Rajeshkumar NV, Garrido-Laguna I, De Oliveira E, Rubio-Viqueira B et al. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer. Mol Cancer Ther 2011; 10: 1311–1316.
Rubio-Viqueira B, Jimeno A, Cusatis G, Zhang X, Iacobuzio-Donahue C, Karikari C et al. An in vivo platform for translational drug development in pancreatic cancer. Clin Cancer Res 2006; 12: 4652–4661.
Stebbing J, Paz K, Schwartz GK, Wexler LH, Maki R, Pollock RE et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer 2014; 120: 2006–2015.
Edgar R, Domrachev M, Lash AE . Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30: 207–210.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5: R80.
Gautier L, Cope L, Bolstad BM, Irizarry RA . affy-analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004; 20: 307–315.
Dai C, Liu J . Inducing pairwise gene interactions from time-series data by EDA based bayesian network. Conf Proc IEEE Eng Med Bio Soc 2005; 7: 7746–7749.
Subramanian J, Simon R . Gene expression-based prognostic signatures in lung cancer: ready for clinical use? J Natl Cancer Inst 2010; 102: 464–474.
Lezhnina K, Kovalchuk O, Zhavoronkov AA, Korzinkin MB, Zabolotneva AA, Shegay PV et al. Novel robust biomarkers for human bladder cancer based on activation of intracellular signaling pathways. Oncotarget 2014; 5: 9022–9032.
Sparks AB, Morin PJ, Vogelstein B, Kinzler KW . Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res 1998; 58: 1130–1134.
Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA . Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer Res 2012; 72: 1547–1556.
Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 1990; 50: 7717–7722.
Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med 2012; 367: 1596–1606.
O'Hara RJ, Greenman J, MacDonald AW, Gaskell KM, Topping KP, Duthie GS et al. Advanced colorectal cancer is associated with impaired interleukin 12 and enhanced interleukin 10 production. Clin Cancer Res 1998; 4: 1943–1948.
Galizia G, Lieto E, De Vita F, Romano C, Orditura M, Castellano P et al. Circulating levels of interleukin-10 and interleukin-6 in gastric and colon cancer patients before and after surgery: relationship with radicality and outcome. J Interferon Cytokine Res 2002; 22: 473–482.
Galizia G, Orditura M, Romano C, Lieto E, Castellano P, Pelosio L et al. Prognostic significance of circulating IL-10 and IL-6 serum levels in colon cancer patients undergoing surgery. Clin Immunol 2002; 102: 169–178.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–2342.
Cassano A, Bagala C, Battelli C, Schinzari G, Quirino M, Ratto C et al. Expression of vascular endothelial growth factor, mitogen-activated protein kinase and p53 in human colorectal cancer. Anticancer Res 2002; 22: 2179–2184.
Yamaguchi T, Bando H, Mori T, Takahashi K, Matsumoto H, Yasutome M et al. Overexpression of soluble vascular endothelial growth factor receptor 1 in colorectal cancer: Association with progression and prognosis. Cancer Sci 2007; 98: 405–410.
Poindessous V, Ouaret D, El Ouadrani K, Battistella A, Megalophonos VF, Kamsu-Kom N et al. EGFR- and VEGF(R)-targeted small molecules show synergistic activity in colorectal cancer models refractory to combinations of monoclonal antibodies. Clin Cancer Res 2011; 17: 6522–6530.
Tabernero J . The role of VEGF and EGFR inhibition: implications for combining anti-VEGF and anti-EGFR agents. Mol Cancer Res 2007; 5: 203–220.
Karapetis CS, Jonker D, Daneshmand M, Hanson JE, O'Callaghan CJ, Marginean C et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer--results from NCIC CTG/AGITG CO.17. Clin Cancer Res 2014; 20: 744–753.
Wu S, Gan Y, Wang X, Liu J, Li M, Tang Y . PIK3CA mutation is associated with poor survival among patients with metastatic colorectal cancer following anti-EGFR monoclonal antibody therapy: a meta-analysis. J Cancer Res Clin Oncol 2013; 139: 891–900.
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11: 753–762.
Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol 2008; 26: 4268–4275.
Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY . Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006; 66: 10100–10111.
van der Veeken J, Oliveira S, Schiffelers RM, Storm G, van Bergen En Henegouwen PM, Roovers RC . Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets 2009; 9: 748–760.
He H, Levitzki A, Zhu HJ, Walker F, Burgess A, Maruta H . Platelet-derived growth factor requires epidermal growth factor receptor to activate p21-activated kinase family kinases. The Journal of biological chemistry 2001; 276: 26741–26744.
Saito Y, Haendeler J, Hojo Y, Yamamoto K, Berk BC . Receptor heterodimerization: essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation. Mol Cell Biol 2001; 21: 6387–6394.
Ware KE, Marshall ME, Heasley LR, Marek L, Hinz TK, Hercule P et al. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS ONE 5: e14117.
Singleton KR, Kim J, Hinz TK, Marek LA, Casas-Selves M, Hatheway C et al. A receptor tyrosine kinase network composed of fibroblast growth factor receptors, epidermal growth factor receptor, v-erb-b2 erythroblastic leukemia viral oncogene homolog 2, and hepatocyte growth factor receptor drives growth and survival of head and neck squamous carcinoma cell lines. Mol Pharmacol 83: 882–893.
Larsen AK, Ouaret D, El Ouadrani K, Petitprez A . Targeting EGFR and VEGF(R) pathway cross-talk in tumor survival and angiogenesis. Pharmacol Ther 131: 80–90.
Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M . Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 140: 268–279.
Saridaki Z, Weidhaas JB, Lenz HJ, Laurent-Puig P, Jacobs B, De Schutter J et al. A let-7 microRNA-binding site polymorphism in KRAS predicts improved outcome in patients with metastatic colorectal cancer treated with salvage cetuximab/panitumumab monotherapy. Clin Cancer Res 2014; 20: 4499–4510.
Luo X, Burwinkel B, Tao S, Brenner H . MicroRNA signatures: novel biomarker for colorectal cancer? Cancer Epidemiol, Biomarker Prev 2011; 20: 1272–1286.
Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA Jr, Sjoblom T et al. The colorectal microRNAome. Proc Natl Acad Sci USA 2006; 103: 3687–3692.
Cappuzzo F, Sacconi A, Landi L, Ludovini V, Biagioni F, D'Incecco A et al. MicroRNA signature in metastatic colorectal cancer patients treated with anti-EGFR monoclonal antibodies. Clin Colorect Cancer 2014; 13: 37–45 e34.
Ruzzo A, Graziano F, Vincenzi B, Canestrari E, Perrone G, Galluccio N et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. The Oncologist 2012; 17: 823–829.
Acknowledgements
This work was supported by NIH grant SPORE P50 DE019032.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zhu, Q., Izumchenko, E., Aliper, A. et al. Pathway activation strength is a novel independent prognostic biomarker for cetuximab sensitivity in colorectal cancer patients. Hum Genome Var 2, 15009 (2015). https://doi.org/10.1038/hgv.2015.9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.9
This article is cited by
-
Rationally co-targeting divergent pathways in KRAS wild-type colorectal cancers by CANscript technology reveals tumor dependence on Notch and Erbb2
Scientific Reports (2017)
-
Using activation status of signaling pathways as mechanism-based biomarkers to predict drug sensitivity
Scientific Reports (2015)

{kind=link}