
Abstract
Idiopathic congenital nystagmus (ICN) is a genetically heterogeneous eye movement disorder that causes a large proportion of childhood visual impairment. Here we describe a missense variant (p.L292P) within a mutation-rich region of FRMD7 detected in three affected male siblings in a Japanese family with X-linked ICN. Combining sequence analysis and results from structural and functional predictions, we report p.L292P as a variant potentially disrupting FRMD7 function associated with X-linked ICN.
Similar content being viewed by others
Idiopathic congenital nystagmus (ICN) is the most common oculomotor disorder, with typical features of bilateral and involuntary oscillations of the eye, visual impairment and abnormal head postures or movement.1–3 The symptoms appear at birth or during the first few months of life. Although the inheritance pattern is heterogeneous, X-linked ICN (XLICN) with incomplete penetrance in females is most common,3,4 and three disease loci of XLICN have been mapped to Xp11.4-p11.3 (NYS5, OMIM# 300589), Xp22.3 (NYS6, OMIM# 300814) and Xq26-q27 (NYS1, OMIM# 310700).1,5,6 Mutations of the four-point-one, ezrin, radixin, moesin (FERM) domain-containing 7 (FRMD7, OMIM#300628) gene, which contains 12 exons and encodes a member of the protein 4.1 superfamily, at Xq26-q27 seem to be the main cause of XLICN in Western and Asian populations.2 Mutations of the G-protein-coupled receptor 143 gene (GPR143, OMIM #300808) at Xp22 are well known to cause ocular albinism (OA) primarily and nystagmus as a secondary phenotype,5 but GPR143 mutations have been also reported in XLICN families, without the classical phenotype of OA.7 In this report, we describe an uncharacterized FRMD7 missense variant (NM_194277.2:c.875T>C) detected in a Japanese family with XLCIN.
The Japanese family included three male siblings affected by nystagmus (Figure 1a, Table 1). The proband (II:3) was born at 39 weeks of gestation as the third child of healthy nonconsanguineous parents. Delay in psychomotor development was apparent during his early infancy. He had horizontal oscillations of both eyes without other ocular abnormalities, which was noted at the age of 2 years and was diagnosed as ICN. His development quotient was 40 as measured by Japanese standard methods at 2 years and 6 months of age, when he was presented to our clinic for physical and speech therapy for his developmental delay. No distinctive features or ophthalmologic abnormality, except nystagmus, was observed on physical examination. The cavum septum pellucidum was detected on performing magnetic resonance imaging, but no other abnormality was detected on performing electroencephalography or auditory brainstem response. His two elder brothers, 12-year-old (II:1) and 10-year-old (II:2), were affected by nystagmus without oscillopsia noted at the age of 11 years and since birth, respectively. Patient II:1 had mild horizontal eye movement and obvious head tilt without myopia, whereas patient II:2 had constant horizontal eye movement and head nodding, with mild myopia, and was surgically treated twice. There were no other ocular or systemic abnormalities, except horizontal and involuntary oscillations of both eyes in both brothers. Although the age of diagnosis was late in II:1 and II:3, nystagmus observed in all three male siblings was diagnosed as an ICN based on clinical features and ophthalmologic findings. Ocular oscillation was not observed in their parents, parents’ siblings or grandparents. Although an autosomal recessive inheritance pattern has been reported in ICN,8 no loci responsible for autosomal recessive ICN have been known so far. All three brothers were affected with ICN, despite unaffected parents; we focused on the ICN inherited in an X-linked inheritance pattern.

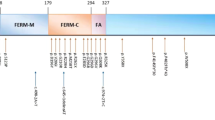
A Japanese family with X-linked congenital nystagmus. (a) Pedigree. The squares and circle represent males and female, respectively. Black symbols indicate affected individuals, and unfilled symbols indicate unaffected individuals. Arrow marks the proband. (b) DNA sequence chromatograms of the FRMD7. Affected family members are denoted by II:1, II:2 and II:3. Arrow marks the variant, c.875T>C. (c) Cross-species multiple alignment of FRMD7 protein sequences around the pL292P variant site, showing evolutionary conservation of the altered residue in the highly conserved residues of the FERM-adjacent (FA) domain. Amino-acid sequence comparison in several related proteins using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The nine proteins depicted are from human, Pan troglodytes, Macaca mulatta, Mus musculus, Rattus norvegicus, Canis familiaris, Bos taurus, Equus caballus and Gallus gallus. The FRMD7 variant, p.L292P, is indicated above the aligned sequence, with the amino acid shaded in the alignment. Arrow heads indicate residues reported to be causative mutations around codon 292 in cases with XLICN.14–20 Gray bars indicate regions of the FERM and FA domains.
The ethical committee of The University of Tokushima approved the study. Informed consent was obtained from all participating family members. Molecular diagnosis was performed using genomic DNA extracted from peripheral blood lymphocytes of the three affected siblings. Mutation analysis of two candidates genes of XLICN, FRMD7 and GPR143, was performed using polymerase chain reaction (PCR) and direct sequencing with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on a 3130 Genetic Analyzer (Applied Biosystems) in both directions, according to published primer sequences covering the sequences of all coding exons and splice junctions.9 Sequencing FRMD7 revealed a T>C transition (c.875T>C) in exon 9 (Figure 1b), which causes a conservative substitution of leucine (L) by proline (P) at codon 292 (p.L292P). This variant co-segregated in all three affected male siblings, although the inheritance of the variant remains unclear because the unaffected mother’s DNA was unavailable (Figures 1a and b). No sequence alteration was observed in GPR143. In proband (II-3), conventional G banding showed a normal male karyotype of 46,XY, and array-based molecular cytogenetic analysis using the HumanOmni1-Quad genotyping array (Illumina, San Diego, CA, USA) and GenomeStudio software (Illumina) revealed no probable causative abnormality for delay in psychomotor development and nystagmus, such as large copy-number alterations, loss of heterozygosity or uniparental disomy, on the basis of suggested guidelines.10,11 We accordingly speculated that the c.875T>C variant of FRMD7 is associated with ICN in three siblings, but it remained unclear whether any genetic alterations including this variant are related to developmental delay observed in the proband (II:3).
The missense variant c.875T>C has a single-nucleotide polymorphism (SNP) number (rs192346335) in the reference SNP database (dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/) but was not found in our 100 unrelated control Japanese males, the 1000 Genomes Project database (http://www.1000genomes.org/) or NHLBI GO Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/). However, this variant was detected in nine individuals in heterozygous state and in one individual in homozygous or hemizygous state among 1102 individuals described in the Japanese genetic variation database (the Human Genetic Variation Database, HGVD; http://www.genome.med.kyoto-u.ac.jp/SnpDB/), although the characteristics of each individual, including sex, are not available in HGVD.
The c.875T>C variant in patients with congenital nystagmus has never been reported previously in databases (Human Gene Mutation Database professional version, http://www.hgmd.cf.ac.uk/ac/index.php and ClinVar, http://www.ncbi.nlm.nih.gov/clinvar/). This variant was reported in one Korean family with ICN, with incomplete penetrance,12 but only limited information without detailed analysis of its pathogenicity was provided in the report. It accordingly remained unclear whether this is a causative variant or a rare benign polymorphism as observed in East Asian population.
We focused on the prediction of effects of p.L292P, which is a highly conserved residue in multiple species (Figure 1c) and lies in the FERM-adjusted (FA) domain between amino acids 288 and 336 (Ensembl, ENSP00000298542), on the structure and function of FRMD7. The FRMD7 protein consists of an N-terminal FERM domain (2–282; ENSP00000298542), a FA domain, and a region with little identity to other proteins.2 Among these regions, the FA domain is one in which mutations are densely concentrated.2 Moreover, >20% of known mutations have been mapped to exon 9, encoding parts of the FERM and FA domains,13 and several causative missense mutations have been identified around codon 292 (Figure 1c).14–20 The effects of the mutations on protein function were assessed by various in silico prediction approaches following the Best Practice Guidelines of the Association for Clinical Genetic Science (http://www.acgs.uk.com/quality-committee/best-practice-guidelines/). The FRMD7 p.L292P mutant protein was predicted with high confidence to be ‘damaging’ by FATHMM v2.3 (score=–2.29; http://fathmm.biocompute.org.uk/), ‘disease causing’ by Mutation Taster (phastCons=4.574 and phyloP=0.998; http://www.mutationtaster.org/), ‘probably damaging’ by Polyphen2 (score=0.993, sensitivity=0.47 and specificity=0.96 in HumVar model; http://genetics.bwh.harvard.edu/pph2/), ‘damaging’ by SIFT (score=0.04; http://sift.jcvi.org/) and ‘pathological’ by Pmut (position-specific independent count score=0.8980; http://mmb2.pcb.ub.es:8080/PMut/). Analysis using Panther (http://www.pantherdb.org/tools/csnpScoreForm.jsp) suggested that the observed amino-acid substitution is functionally deleterious (substitution position-specific evolutionary conservation score=–7.03876). In addition, using the Phyre2 program (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index), we predicted the secondary structures of the wild- and mutant-type FRMD7 proteins. The change of amino acid in the mutant protein led to a loss of the secondary structure in the helix around the L292 residue (Supplementary Figure 1).
Taking these lines of evidence together, p.L292P appears to exert a marked effect on FRMD7 protein functionality and to be a variant responsible for XLICN, rather than a benign polymorphism. Given that incomplete penetrance and variable expression of XLICN have been observed in females who carry mutations in the FRMD7,2,20 it is not surprising that nine individuals (probably females) in heterozygous state were observed among 1,102 individuals analyzed in the Japanese genetic variation database (HGVD). Although the identified FRMD7 mutations are scattered over almost all exons and splice sites,2 there are mutation-rich exons/regions, and these should be treated as the most important candidate regions when screening for mutations is performed. Predictive analyses of the structure and function of possible mutant proteins observed in candidate regions predict the consequences of mutations,13,20 and provide meaningful improvement of clinical genetic diagnosis. Further functional and/or biochemical studies of FRMD7 mutations will shed light on the molecular mechanisms underlying the pathogenesis of XLICN, which are not yet fully understood.2
References
References
Cabot A, Rozet JM, Gerber S, Perrault I, Ducroq D, Smahi A et al. A gene for X-linked idiopathic congenital nystagmus (NYS1) maps to chromosome Xp11.4–p11.3. Am J Hum Genet 1999; 64: 1141–1146.
Watkins RJ, Thomas MG, Talbot CJ, Gottlob I, Shackleton S . The Role of FRMD7 in Idiopathic Infantile Nystagmus. J Ophthalmol 2012; 2012: 460956.
Self J, Lotery A . The molecular genetics of congenital idiopathic nystagmus. Semin Ophthalmol 2006; 21: 87–90.
Thomas S, Proudlock FA, Sarvananthan N, Roberts EO, Awan M, McLean R et al. Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain 2008; 131: 1259–1267.
Bassi MT, Schiaffino MV, Renieri A, De Nigris F, Galli L, Bruttini M et al. Cloning of the gene for ocular albinism type 1 from the distal short arm of the X chromosome. Nat Genet 1995; 10: 13–19.
Kerrison JB, Vagefi MR, Barmada MM, Maumenee IH . Congenital motor nystagmus linked to Xq26–q27. Am J Hum Genet 1999; 64: 600–607.
Liu JY, Ren X, Yang X, Guo T, Yao Q, Li L et al. Identification of a novel GPR143 mutation in a large Chinese family with congenital nystagmus as the most prominent and consistent manifestation. J Hum Genet 2007; 52: 565–570.
Abadi RV, Bjerre A . Motor and sensory characteristics of infantile nystagmus. Br J Ophthalmol 2002; 86: 1152–1160.
Yan N, Liao X, Cai SP, Lan C, Wang Y, Zhou X et al. A novel nonsense mutation of the GPR143 gene identified in a Chinese pedigree with ocular albinism. PLoS ONE 2012; 7: e43177.
Kearney HM, Thorland EC, Brown KK, Quintero-Rivera F, South ST, Working Group of the American College of Medical Genetics Laboratory Quality Assurance Committee. American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med 2011; 13: 680–685.
Kearney HM, Kearney JB, Conlin LK . Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 2011; 31: 595–613.
Oh SY, Shin BS, Seo MW, Ki CS, Hwang JM, Kim JS . Novel mutation in FRMD7 gene in X-linked congenital nystagmus. J Korean Bal Soc 2007; 6: 155–160 (Korean).
Zhu Y, Zhuang J, Ge X, Zhang X, Wang Z, Sun J et al. Identifcation of a novel mutation p.I240T in the FRMD7 gene in a family with congenital nystagmus. Sci Rep 2013; 3: 3084.
Zhang B, Liu Z, Zhao G, Xie X, Yin X, Hu Z et al. Novel mutations of the FRMD7 gene in X-linked congenital motor nystagmus. Mol Vis 2007; 13: 1674–1679.
Tarpey P, Thomas S, Sarvananthan N, Mallya U, Lisgo S, Talbot CJ et al. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet 2006; 38: 1242–1244.
He X, Gu F, Wang Y, Zhang L, Dai S, Li H et al. A novel mutation in FRMD7 causing X-linked idiopathic congenital nystagmus in a large family. Mol Vis 2008; 14: 56–60.
Li N, Wang L, Cui L, Zhang L, Dai S, Li H et al. Five novel mutations of the FRMD7 gene in Chinese families with X-linked infantile nystagmus. Mol Vis 2008; 14: 733–738.
Schorderet DF, Tiab L, Gaillard MC, Lorenz B, Klainguti G, Kerrison JB et al. Novel mutations in FRMD7 in X-linked congenital nystagmus. Hum Mutat 2007; 28: 525.
Hu Y., Shen J, Zhang S, Yang T, Huang S, Yuan H . A novel splicing mutation of the FRMD7 gene in a Chinese family with X-linked congenital nystagmus. Mol Vis 2012; 18: 87–91.
Radhakrishna U, Ratnamala U, Deutsch S, Bartoloni L, Kuracha MR, Singh R et al. Novel homozygous, heterozygous and hemizygous FRMD7 gene mutations segregated in the same consanguineous family with congenital X-linked nystagmus. Eur J Hum Genet 2012; 20: 1032–1036.
Data Citations
Imoto, Issei HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.572 (2015)
Acknowledgements
The authors thank the family members as well as the healthy volunteers for their enthusiasm and participation in this study. This work was supported by JSPS KAKENHI Grant Numbers 60645216 (NO) and 26293304 (II) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplemental Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Kohmoto, T., Okamoto, N., Satomura, S. et al. A FRMD7 variant in a Japanese family causes congenital nystagmus. Hum Genome Var 2, 15002 (2015). https://doi.org/10.1038/hgv.2015.2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.2
