
Abstract
We report a Japanese family with spastic paraplegia 7 (SPG7) that carries a deleterious homozygous p.R398X mutation in SPG7. The patients showed a predominant cerebellar ataxia phenotype. SPG7 is quite rare in Japan, but it should be included in the differential diagnosis for hereditary spastic-ataxic syndromes, even if the cerebellar signs are much more pronounced than the pyramidal tract signs.
Similar content being viewed by others
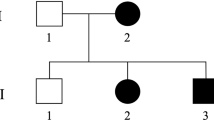


Hereditary ataxias and hereditary spastic paraplegias (HSPs) are a group of clinically and genetically heterogeneous neurodegenerative disorders.1–3 The predominant feature of HSPs is progressive lower limb spasticity and weakness caused by the degeneration of corticospinal tracts. HSPs are often associated with additional neurological and systemic features such as cognitive impairment, cerebellar ataxia, peripheral neuropathy and hematologic and skin abnormalities; these HSP syndromes are referred to as complicated HSP.2,3 Because of more than 70 genetic loci, it is quite problematic to make a correct diagnosis of a particular genetic form of HSP. In particular, the combined presence of pyramidal features and ataxia, so-called spastic-ataxic syndromes, is troublesome for the differential diagnosis.4 Many clinicians often struggle to decide whether the patient has a hereditary ataxia with pyramidal tract signs or HSP with cerebellar ataxia.
Here, we report a family with spastic paraplegia 7 (SPG7) showing the phenotype of a nearly pure cerebellar ataxia with less predominant spasticity. This family had been followed as a certain subtype of hereditary ataxias. Whole-exome sequencing (WES) identified a homozygous p.R398X mutation in SPG7 in this family. To our knowledge, this is the first report of the p.R398X mutation in SPG7 in the Japanese population and this family is the second published one with SPG7 in Japan.5
The proband (case 1) was a 74-year-old man with hypertension. His parents were reported not to be consanguineous. His mother and father died at age 42 and 80, respectively, and they were free of neurological symptoms in their lifetime. At age 32, the proband began to note staggering upon walking. He developed a hand tremor during writing and dysarthria at age 41 and then pollakiuria at age 45. When he first visited a neurology clinic at age 47, he presented with dysarthria, limb ataxia, exaggerated deep tendon reflexes without extensor planter responses and ataxic gait. Head computed tomography revealed a mild cerebellar hemispheric atrophy. His condition was diagnosed as spinocerebellar degeneration.
Thereafter, his ataxic gait progressed slowly, and he became wheelchair bound at age 52. At his latest visit at age 74, his mini-mental state examination score was 30. He had no optic nerve atrophy, ophthalmoplegia, nystagmus or blepharoptosis. The deep tendon reflexes were increased in all four limbs, but Babinski sign was not elicited bilaterally. Neither ankle nor knee clonus was observed. He had no involuntary movements, rigidity or bradykinesia. Truncal and limb ataxia appeared exacerbated. He showed a marked staggering and could not walk without holding on to something. His scores on Scale for the Assessment and Rating of Ataxia and International Co-operative Ataxia Rating Scale were 23 and 58, respectively. He complained of pollakiuria and urinary incontinence but not of constipation or orthostatic hypotension. There was no evidence of sensory disturbance. His condition was still consistent with cerebellar ataxia associated with urinary disturbance. A nerve conduction study revealed no signs of neuropathy. Brain magnetic resonance imaging showed an atrophy in the cerebellar hemisphere but not in the brainstem, corpus callosum or cerebrum (Figure 1).

(a) MR images of the patients (case 1: 74-years old; case 2: 71-years old). Atrophy is observed in the cerebellar hemisphere but not in the cerebellar vermis, brainstem or corpus callosum, in both patients. The hot cross bun sign is not observed. (b) Sanger sequencing. A homozygous c.1192C>T/p.R398X mutation in SPG7 is indicated (arrows) (upper panel: case 1; lower panel: case 2).
The younger brother of the proband (case 2) first noticed gait disturbance with a feeling of shaking knees at age 50. At age 51, he developed dysarthria and urinary dysfunction. Neurological examination at age 52 showed ataxic dysarthria, exaggerated patellar and Achilles tendon reflexes without Babinski signs and lower limb ataxia. His gait was wide-based and staggering. He started to complain of a sense of stiffness in the lower limbs at age 55. His gait disturbance gradually worsened, and he became dependent on a cane at age 66. At his latest visit at age 71, his mini-mental state examination score was 29. There was no optic nerve atrophy. He showed a limitation in horizontal eye movement, but no blepharoptosis or nystagmus was noted. Ataxic dysarthria was evident. His deep tendon reflexes were normal in the upper limbs but enhanced in the lower limbs, and Babinski sign was positive bilaterally. Extrapyramidal signs were not noted. His gait was wide-based, staggering and spastic. His scores of Scale for the Assessment and Rating of Ataxia and International Co-operative Ataxia Rating Scale were 20 and 41, respectively. He experienced pollakiuria but neither constipation nor orthostatic hypotension. He showed no signs of sensory disturbance, and a nerve conduction study was not particular. He had a slight talipes equinovarus deformity bilaterally. His brain magnetic resonance imaging findings were essentially the same as his elder brother (Figure 1).
The elder sister of the proband (case 3) was reported to develop gait disturbance at age 50. She was diagnosed with spinocerebellar degeneration, but the clinical details were not available because she died prior to this study. Based on the positive family history and the clinical and neuroradiological findings, these patients were suspected as having a certain subtype of hereditary ataxias, although the family history was not fully informative on whether the mode of inheritance was autosomal dominant or recessive.
After obtaining informed consents from cases 1 and 2, we performed genetic testing; autosomal dominant cerebellar ataxias including SCA1, SCA2, MJD/SCA3, SCA6, SCA7, SCA12, SCA17, DRPLA and SCA31 were ruled out. Then, WES was performed for case 1 as previously described.6 Briefly, genomic DNA was captured by using a SureSelect Human All Exon v5 kit (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a HiSeq2000 with 101 bp paired-end reads (Illumina, San Diego, CA, USA). Image analysis and base calling were performed by sequence control software real-time analysis and CASAVA software v1.8 (Illumina). The reads were aligned to GRCh37 with Novoalign (http://www.novocraft.com/). PCR duplicates were removed using Picard (http://picard.sourceforge.net/). Variants were called by the Genome Analysis Toolkit (http://www.broadinstitute.org/gatk/) and annotated by using ANNOVAR (http://www.openbioinformatics.org/annovar/) after excluding the common variants registered in the common dbSNP135 database (minor allele frequency ⩾0.01). The WES mean depth of coverage against the RefSeq coding sequence was 71.8x, and 86.7% of the total coding sequence bases were covered by 20 reads or more. We obtained a rare protein-altering and splice-site variant calls after filtering against 575 in-house control exomes. WES revealed a homozygous c.1,192C>T/p.R398X mutation in SPG7. This mutation was not detected in 575 control exomes, supporting that the carrier frequency of this mutation is quite rare in the Japanese population. Sanger sequencing strongly supported that both patients were homozygotes for this mutation (Figure 1). More precisely, however, the possibility could not be completely ruled out that the patients were compound heterozygotes of the p.R398X mutation and an unidentified large deletion involving SPG7.
SPG7 encodes paraplegin, a 795-amino acid protein that is a subunit of the m-AAA (ATPases associated with diverse cellular activities) protease.7,8 The m-AAA protease, which is composed of paraplegin and AFG3L2, is involved in protein quality control in the mitochondrial inner membrane. It is very likely that the p.R398X mutation causes nonsense-mediated RNA decay. The truncated protein, even if it is present, might result in drastic structural and functional defects of the complex because the truncation occurs in the AAA domain of paraplegin.
SPG7 usually manifests as complicated HSP syndrome and is frequently associated with cerebellar ataxia.2,3,9 Both affected individuals (cases 1 and 2) presented with a marked cerebellar ataxia with less prominent spasticity. They did not show any cognitive impairment, optic atrophy or peripheral neuropathy. It is quite difficult to make a correct diagnosis for a particular subtype of ARCAs or autosomal dominant cerebellar ataxias in the case of a predominant cerebellar phenotype of adult onset without extracerebellar or non-central nervous system symptoms. Our cases were roughly included in this category, and therefore, we conducted WES analysis and reached a final diagnosis of SPG7.
Our cases indicate that SPG7 can manifest as a nearly pure cerebellar ataxia phenotype. Both patients complained of urinary frequency, but detailed urodynamic studies were not performed. Retrospectively, bladder dysfunction might be suggestive of SPG7 because approximately half of the patients (22/42) exhibited bladder dysfunction in a large Dutch cohort including 60 SPG7 patients.10
One report described a sporadic SPG7 patient who was a compound heterozygote of the p.R398X/p.A510V mutation in SPG7, which is the most common SPG7 mutation in Caucasian countries.11 He demonstrated cerebellar ataxia, but the brain magnetic resonance imaging was normal. In a large Dutch cohort described above, cerebellar ataxia was observed in 57% (27/47) of the patients.10 The authors suggested that the cerebellar phenotype of SPG7 might be correlated with SPG7 null alleles.10 Our observation is consistent with this speculation.
In conclusion, we report here that SPG7 can present with the predominant cerebellar phenotype masquerading as SCA6 or cortical cerebellar atrophy. In Japan, the prevalence of SPG7 is considered very rare,12 but it needs to be considered in the differential diagnosis of hereditary spastic-ataxic syndromes, even if the cerebellar signs are much more predominant than the pyramidal tract signs.
References
References
Jayadev S, Bird TD . Hereditary ataxias: overview. Genet Med 2013; 15: 673–683.
Fink JK . Hereditary spastic paraplegia: clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol 2013; 126: 307–328.
Finsterer J, Löscher W, Quasthoff S, Wanschitz J, Auer-Grumbach M, Stevanin G . Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 2012; 318: 1–18.
de Bot ST, Willemsen MAAP, Vermeer S, Kremer HPH, van de Warrenburg BPC . Reviewing the genetic causes of spastic-ataxias. Neurology 2012; 79: 1507–1514.
Doi H, Ohba C, Tsurusaki Y, Miyatake S, Miyake N, Saitsu H et al. Identification of a novel homozygous SPG7 mutation in a Japanese patient with spastic ataxia: making an efficient diagnosis using exome sequencing for autosomal recessive cerebellar ataxia and spastic paraplegia. Intern Med 2013; 52: 1629–1633.
Yoshida K, Miyatake S, Kinoshita T, Doi H, Tsurusaki Y, Miyake N et al. ‘Cortical cerebellar atrophy’ dwindles away in the era of next-generation sequencing. J Hum Genet 2014; 59: 589–590.
Rugarli EI, Langer T . Translating m-AAA protease function in mitochondria to hereditary spastic paraplegia. Trends Mol Med 2006; 12: 262–269.
Martinelli P, Rugarli EI . Emerging roles of mitochondrial proteases in neurodegeneration. Biochim Biophys Acta 2010; 1797: 1–10.
Elleuch N, Depienne C, Benomar A, Hernandez AM, Ferrer X, Fontaine B et al. Mutation analysis of the paraplegin gene (SPG7) in patients with hereditary spastic paraplegia. Neurology 2006; 66: 654–659.
van Gassen KL, van der Heijden CD, de Bot ST, den Dunnen WF, van den Berg LH, Verschuuren-Bemelmans CC et al. Genotype-phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain 2012; 135: 2994–3004.
Schlipf NA, Schüle R, Klimpe S, Karle KN, Synofzik M, Schicks J et al. Amplicon-based high-throughput pooled sequencing identifies mutations in CYP7B1 and SPG7 in sporadic spastic paraplegia patients. Clin Genet 2011; 80: 148–160.
Ishiura H, Takahashi Y, Hayashi T, Saito K, Furuya H, Watanabe M et al. Molecular epidemiology and clinical spectrum of hereditary spastic paraplegia in the Japanese population based on comprehensive mutational analyses. J Hum Genet 2014; 59: 163–172.
Data Citations
Yoshida, Kunihiro HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.580 (2015)
Acknowledgements
This study was supported in part by a grant from the Research Committee for Ataxic Diseases, the Ministry of Health, Labor and Welfare, Japan (KY). This study was also supported by the Takeda Science Foundation (NM); the Yokohama Foundation for Advancement of Medical Science (SM); the Japan Society for the Promotion of Science (a Grant-in-Aid for Scientific Research (C) (SM), and a Grant-in-Aid for Scientific Research (A) (NM)); the Strategic Research Program for Brain Sciences; and a Grant-in-Aid for Scientific Research on Innovative Areas (Transcription Cycle) (NM) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Yahikozawa, H., Yoshida, K., Sato, S. et al. Predominant cerebellar phenotype in spastic paraplegia 7 (SPG7). Hum Genome Var 2, 15012 (2015). https://doi.org/10.1038/hgv.2015.12
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.12
