
Abstract
We present here a case of attenuated familial adenomatous polyposis (AFAP) with a family history of desmoids and thyroid tumors. This patient had no colonic polyps but did have multiple desmoids. Genetic analysis identified a 4-bp deletion in codon 2644 (c.7932_7935delTTAT: p.Tyr2645LysfsX14) of the adenomatous polyposis coli (APC) gene. In cases with limited numbers of colonic polyps and desmoids, AFAP may be caused by a mutation in the 3′ region of APC.
Similar content being viewed by others
Familial adenomatous polyposis (FAP) is an inherited disorder characterized by the early onset of multiple adenomatous polyps in the colon and rectum. Unless the colon is removed, most patients with this syndrome develop colorectal cancer by the age of 35–40 years. Gardner’s syndrome is a variant of FAP characterized by colonic polyposis, osteomas and soft tissue tumors, such as desmoids. Desmoid tumors are benign, non-inflammatory fibroblastic neoplasms that tend to be locally invasive but are not metastatic. However, they have a high potential for local recurrence following surgical resection. Upper gastrointestinal neoplasms are also typical extracolonic manifestations in FAP. Histologically, the majority of duodenal polyps are adenomas, whereas the majority of gastric polyps are fundic gland polyps. Similar to other hereditary cancer syndromes, there is an increased risk of additional malignancies in FAP, including thyroid cancer.
Germline mutations of the adenomatous polyposis coli (APC) gene that is responsible for FAP may also cause Gardner’s syndrome. Correlations have been observed between mutation in APC and the FAP phenotype. Mutations located between codons 1250 and 1464, especially those at codon 1309, have been reported to be correlated with the development of profuse-type polyposis (>5,000 colorectal polyps).1,2 This region is closely associated with the mutation cluster region (codons 1286–1513), where more than 60% of somatic mutations occurring in colon cancer patients are clustered.3 Attenuated FAP (AFAP) is a variant of FAP that is characterized by the presence of less than 100 polyps with a more proximal colonic location in addition to a delayed age of colorectal cancer onset. Mutations in AFAP patients have been reported to be located in three distinct regions of the APC gene, including the 5′ end spanning exons 3 to 5, exon 9 and the 3′ distal end.4 Previous studies of desmoid tumor patients have revealed that patients with mutations beyond codon 1444, especially between codons 1445 and 1578, develop desmoid tumors more frequently than those with mutations in other regions of the APC gene.5,6
Here, we report a case of a desmoid tumor patient with a family history of desmoid tumors, thyroid cancer and colonic polyposis. Using a next-generation sequencing method, we identified a 4-bp deletion in the carboxyl terminus of the APC gene (c.7932_7935delTTAT: p.Tyr2645LysfsX14). Our findings provide evidence that the deletion of the 3′ distal end of the APC protein impairs its function of suppressing desmoid tumors and thyroid cancer, thereby deepening the current understanding of the correlation between the phenotype and genotype of FAP.
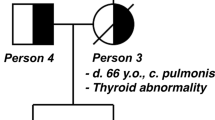
A 27-year-old female with desmoid tumors in front of the sternum and on the left elbow visited our hospital for genetic counseling. Evaluation of her family history revealed that her father underwent total colectomy for multiple colon polyps, resection of two abdominal desmoid tumors at the age of 50 years and resection of thyroid and prostate cancer at 52 years of age (Figure 1a). A paternal aunt also underwent resection of several desmoid tumors on the back and developed thyroid cancer in her forties (Figure 1a). Furthermore, the paternal grandfather suffered from colon cancer with multiple colon polyps and cancer of the duodenal papilla (Figure 1a). Colonoscopy showed that the patient had no colorectal polyps. However, gastroscopy revealed numerous fundic gland polyps in her stomach. Considering these findings, we diagnosed her with AFAP.

Identification of an APC mutation in a pedigree of AFAP with desmoid tumors. (a) The pedigree of the family assessed in this study. Open square: normal male individual; closed square: affected male individual; open circle: normal female individual; closed circle: affected female individual; slash: deceased individual; and arrow: proband. (b) Validation of the 4-bp deletion in APC. Direct sequencing of the PCR products of DNA from a healthy volunteer and a patient revealed a deletion of TTAT at codon 2644 of the APC gene. The amino acid code and position are shown. The red square indicates the four nucleotides deleted in the patient’s DNA. AFAP, attenuated familial adenomatous polyposis; APC, adenomatous polyposis coli.
After obtaining written informed consent from the patient, genomic DNA was extracted from her peripheral blood leucocytes. We first performed the direct sequencing of APC to examine the 5′ half of the coding region, which is where most APC mutations occur. However, no pathogenic mutations were detected by this initial examination. Although examinations of other regions, including the remaining 3′ region and the promoter region, structural analysis of APC by multiplex ligation-dependent probe amplification and the sequencing of other polyposis-related genes, such as MUTYH, POLD and POLE, by the Sanger method, were possible for the second screening, we conducted whole-genome sequencing to examine the utility of next-generation sequencing in clinical testing. A library of approximately 550 bp constructed from 200 ng of genomic DNA was prepared and subsequently sequenced using HiSeq 2500 platforms according to the manufacturer's instructions (Illumina, San Diego, CA, USA).
As a result, seven variants were detected in the coding region of APC. Of note, six of the seven variants were detected in the initial screening by direct sequencing using the Sanger method, and no other variants were identified. Among the six variants, five were synonymous single nucleotide variants and one was a nonsynonymous single nucleotide variant (Table 1), as determined by evaluations using public databases, including dbSNP (in NCBI),7 the HapMap project8 and the Human Genetic Variation Browser.9 The remaining variant detected by whole-genome sequencing was a 4-bp deletion of nucleotides 7932 to 7935, c.7932_7935delTTAT: p.Tyr2645LysfsX14 (Table 1, Figure 1b), located outside of the region that we had initially screened. This variant causes a frame shift, leading to a premature stop codon near the 3′ end of APC. With regard to other polyposis-related genes, including MUTYH, POLD1 and POLE, no pathogenic alterations and two single nucleotide polymorphisms (rs3219489 and rs5744751) were detected in these three genes.
APC mutations associated with the incidence of desmoids reportedly accumulate within codons 1445–1578.5,6 In contrast, the mutation identified here was located at codon 2644, within the 3′ region of APC. An identical mutation has been deposited in the International Society for Gastrointestinal Hereditary Tumours (InSiGHT) Variant Databases and has been reported on two different pedigrees.10,11 Although no detailed clinical information was supplied for one of the two patients with this mutation,11 the family history of the other showed that four of six affected family members developed desmoid tumors.10 The patient evaluated in this study also had a family history of desmoid tumors. Notably, the father and aunt in our case developed thyroid cancer, although histological data of their tumors are not available. These findings suggest that this mutation impairs the functioning of the APC product that is responsible for the suppression of desmoids and thyroid cancer.
The APC gene product possesses several domains that are critical for its function. The most prominent function of APC is its ability to regulate Wnt/β-catenin-mediated gene transcription. Mutations in the mutation cluster region lead to the loss of the ability to suppress β-catenin-mediated signaling, which plays a crucial role in the development of colorectal adenoma. The mutant APC protein identified in this study is assumed to have a domain that interacts with β-catenin, which may account for her lack of adenomatous polyps. Reportedly, other mutations located within the 3′ region of APC may also lead to the development of desmoid tumors.12,13 Interestingly, the accumulation of β-catenin has been observed in desmoid tumors of a patient with a mutation at the 3′ end of APC. 14 Thus, a second hit in the wild-type APC allele may suppress the function of the truncated APC protein containing the β-catenin-interacting domain. Because the mutation in this case leads to the truncation of approximately 200 C-terminal amino acids, the mutant protein likely lost domains for interacting with EB1 and human homolog of Drosophila discs large (hDLG).15,16 The APC-EB1 and APC-hDLG complexes are reportedly important for the regulation of chromosome segregation and the cell cycle, respectively.17,18 Therefore, the loss of interaction of this APC product with EB1 and hDLG might cause the deregulation of cell division and proliferation.
With regard to thyroid cancer in FAP, the genotype–phenotype correlation is controversial. Recently, Septer et al.19 have reported an increased risk of thyroid cancer in FAP patients with a mutation at the 5′ end (proximal to codon 528) and at codon 1061 of APC. Of note, this group has also reported two patients with an APC mutation at codon 2092 who developed thyroid cancer. Therefore, mutations at the 3′ end may also increase the risk of thyroid cancer. As more data are collected, these will help to elucidate the genotype–phenotype correlation of thyroid cancer in FAP.
In the family evaluated in this report, neither of the two females with desmoids had colonic polyps, but both of the affected males had colonic polyposis. This intrafamilial phenotypic variation might be attributed to influences of modifier genes or environmental factors.
In conclusion, we identified a rare APC mutation in a desmoid tumor patient with a family history of AFAP with desmoids and thyroid cancer. This mutation results in the production of an APC protein with a truncation at the C-terminal distal end, which renders it incapable of suppressing the development of desmoids and thyroid cancer. In the diagnosis of AFAP patients with desmoids, mutation screening of not only the 5′ region but also of the 3′ region of the APC gene is necessary.
References
References
Bertario L, Russo A, Sala P, Varesco L, Giarola M, Mondini P et al. Multiple approach to the exploration of genotype-phenotype correlations in familial adenomatous polyposis. J Clin Oncol 2003; 21: 1698–1707.
Nagase H, Miyoshi Y, Horii A, Aoki T, Ogawa M, Utsunomiya J et al. Correlation between the location of germ-line mutations in the APC gene and the number of colorectal polyps in familial adenomatous polyposis patients. Cancer Res 1992; 52: 4055–4057.
Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992; 1: 229–233.
Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S et al. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 1998; 62: 1290–1301.
Caspari R, Olschwang S, Friedl W, Mandl M, Boisson C, Boker T et al. Familial adenomatous polyposis: desmoid tumours and lack of ophthalmic lesions (CHRPE) associated with APC mutations beyond codon 1444. Hum Mol Genet 1995; 4: 337–340.
Davies DR, Armstrong JG, Thakker N, Horner K, Guy SP, Clancy T et al. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene. Am J Hum Genet 1995; 57: 1151–1158.
Smigielski EM, Sirotkin K, Ward M, Sherry ST . dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res 2000; 28: 352–355.
International HapMap C, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861.
Narahara M, Higasa K, Nakamura S, Tabara Y, Kawaguchi T, Ishii M et al. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS One 2014; 9: e100924.
Brensinger JD, Laken SJ, Luce MC, Powell SM, Vance GH, Ahnen DJ et al. Variable phenotype of familial adenomatous polyposis in pedigrees with 3′ mutation in the APC gene. Gut 1998; 43: 548–552.
Miyoshi Y, Ando H, Nagase H, Nishisho I, Horii A, Miki Y et al. Germ-line mutations of the APC gene in 53 familial adenomatous polyposis patients. Proc Natl Acad Sci USA 1992; 89: 4452–4456.
Eccles DM, van der Luijt R, Breukel C, Bullman H, Bunyan D, Fisher A et al. Hereditary desmoid disease due to a frameshift mutation at codon 1924 of the APC gene. Am J Hum Genet 1996; 59: 1193–1201.
Scott RJ, Froggatt NJ, Trembath RC, Evans DG, Hodgson SV, Maher ER . Familial infiltrative fibromatosis (desmoid tumours) (MIM135290) caused by a recurrent 3′ APC gene mutation. Hum Mol Genet 1996; 5: 1921–1924.
Couture J, Mitri A, Lagace R, Smits R, Berk T, Bouchard HL et al. A germline mutation at the extreme 3' end of the APC gene results in a severe desmoid phenotype and is associated with overexpression of beta-catenin in the desmoid tumor. Clin Genet 2000; 57: 205–212.
Matsumine A, Ogai A, Senda T, Okumura N, Satoh K, Baeg GH et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science 1996; 272: 1020–1023.
Su LK, Burrell M, Hill DE, Gyuris J, Brent R, Wiltshire R et al. APC binds to the novel protein EB1. Cancer Res 1995; 55: 2972–2977.
Draviam VM, Shapiro I, Aldridge B, Sorger PK . Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J 2006; 25: 2814–2827.
Ishidate T, Matsumine A, Toyoshima K, Akiyama T . The APC-hDLG complex negatively regulates cell cycle progression from the G0/G1 to S phase. Oncogene 2000; 19: 365–372.
Septer S, Slowik V, Morgan R, Dai H, Attard T . Thyroid cancer complicating familial adenomatous polyposis: mutation spectrum of at-risk individuals. Hered Cancer Clin Pract 2013; 11: 13.
Data Citations
Furukawa, Yoichi HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.578 (2015)
Acknowledgements
This work was supported in part by a Grant-in-Aid from the Center of Innovation program of the Japan Science and Technology Agency.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ikenoue, T., Yamaguchi, K., Komura, M. et al. Attenuated familial adenomatous polyposis with desmoids caused by an APC mutation. Hum Genome Var 2, 15011 (2015). https://doi.org/10.1038/hgv.2015.11
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.11
