
Abstract
Osteogenesis imperfecta IIC (OI IIC), which is a rare variant of lethal OI that has been considered to be an autosomal recessive trait, is characterized by twisted, slender long bones with dense metaphyseal margins. Here, we report a typical case of OI IIC caused by a novel heterozygous mutation in the C-propeptide region of COL1A1. OI IIC seems to be caused by a dominant mutation of COL1A1.
Similar content being viewed by others


Osteogenesis imperfecta (OI) comprises a heterogeneous group of connective tissue disorders characterized by fragile bones with susceptibility to fractures. Currently, a molecular genetic classification of OI contains 15 types which display either autosomal-dominant or autosomal-recessive patterns of inheritance and exhibit broad variations in clinical severity (OI type I-XV; MIM #166200, #166210, #259420, #166220, #610967, #610968, #610682, #610915, #259440, #613848, #613849, #613982, #614856, #615066 and #615220).1
Most cases of OI are caused by heterozygous mutations in COL1A1 or COL1A2 (OI type I–IV), the genes encoding the two type I procollagen alpha chains, proα1 (I) and proα2 (I). Each chain contains a core triple helical domain, composed of uninterrupted repeats of the Gly–Xaa–Yaa tripeptide, flanked by propeptides at both the amino- and carboxyl-terminal ends. From the practical viewpoint, OI is clinically diagnosed using the Sillence classification,2 according to which lethal OI due to COL1A1/A2 mutations are classified as types IIA, IIB and IIC. Among these three type II OI, OI IIC is extremely rare, with distinct phenotypical and radiological manifestations, such as slender, twisted long bones with fractures and normal height of the vertebral bodies.3–5 Slender, twisted long bones contrast with thick, crumpled ones seen in other forms of lethal OI, whereas normal height of the vertebral bodies differs from multiple compression fractures of the vertebral bodies in other lethal OI. Another hallmark of OI IIC is osteosclerosis in the metaphyseal ends, outer margins of the flat bones and fracture surfaces.
OI IIC had been believed to be inherited as an autosomal recessive trait, based on affected individuals in the same kindred.4,6 Pace et al.7 reported a case of lethal OI caused by a heterozygous mutation (D1441Y) in the C-propeptide region of COL1A1 They described the phenotype as a lethal variant of OI with features of dense bone diseases. However, radiological examination showed that the phenotype was consistent with that of OI IIC. Moreover, we have previously reported that heterozygous C-propeptide mutations of COL1A1 are responsible for OI IIC (c.4247delC), with a phenotype similar to but slightly milder than OI IIC (A1387V).8 The heterozygosity of these cases contradicted the initial hypothesis that OI IIC was inherited as an autosomal recessive trait. However, the conclusion was not decisive because of the limited number of cases. Here, we report another case with a typical OI IIC phenotype caused by a novel heterozygous frameshift mutation in the C-propeptide region of COL1A1. This observation enhances the hypothesis of the pathogenic link between OI IIC and the C-propeptide heterozygous mutation in COL1A1.
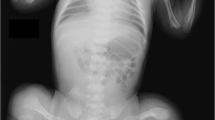
The patient was a male child with healthy parents. Prenatal ultrasonography at 20 weeks of gestation showed short limbs and a hypoplastic bell-shaped thorax. The pregnancy was electively interrupted at 21 weeks’ gestation. Disproportionately short and bent limbs, a hypoplastic thoracic cage and caput membranaceum were noted. The findings of post-mortem radiography fulfilled the radiological criteria of OI IIC, including slender ribs with multiple fractures, erratic ossifications of the flat bones, slender and twisted long bones with sclerosis of the metaphyseal ends and fracture surfaces and normal height of the vertebral bodies (Figure 1).

Post-mortem radiographs of patient. The findings of post-mortem radiography fulfilled the radiological criteria of osteogenesis imperfecta IIC, including slender ribs with multiple fractures, erratic ossifications of the flat bones, slender and twisted long bones with sclerosis of the metaphyseal ends and fracture surfaces and normal height of the vertebral bodies.
After genetic counseling and with written informed consent, we obtained genomic DNA from the umbilical cord blood (patient) and peripheral blood (parents) by using a standard technique. This study was approved from the institutional review board of the Keio University School of Medicine. We checked all the coding exons and flanking introns of COL1A1 and COL1A2 by PCR and direct sequencing. Deletion in and duplications of COL1A1 and COL1A2 were examined by multiplex ligation-dependent probe amplification (MLPA) analyses (SALSA MLPA KIT P271, P272; MRC-Holland, Amsterdam, The Netherlands). We found a novel heterozygous mutation in COL1A1, c.4309delC (p.L1437CfsX89), located in the C-propeptide region of pro α1 (I) (Figure 2). The mRNA with c.4309delC seems to escape the nonsense-mediated decay, because this frameshift occurrs in the last exon of COL1A1. The unaffected parents did not have this mutation. MLPA analysis did not detect exon-level deletion or duplication. The p.L1437CfsX89 was not detected in 150 healthy Japanese controls and was absent from database, including dbSNP, the 1,000 Genomes Project, Exome Variant Server, NHLBI Exome Sequencing Project and the Human Genetic Variation Database (HGVD) in Japanese.

Identification of mutation in the C-propeptide region of COL1A1. Partial sequences of PCR products of the patient and control are shown. Heterozygous single base pair deletion (c.4309delC) in the patient is indicated by arrows.
Here we describe the fourth observation of OI IIC with a C-propeptide mutation in COL1A1. The histological and radiological observations indicate that the distinctive skeletal changes in OI IIC or the ‘dense bone phenotype’ is related to the abundance of immature woven bone in the skeleton.7,8 However, the phenotypic consequences of C-propeptide mutations of COL1A1 reported thus far have ranged from mild OI type I to lethal OI type II.7–12 Thus, the type of C-propeptide mutations responsible for the dense bone phenotype observed in OI IIC remain elusive. The absence of vertebral compression fracture in OI IIC deserve comment. Histologic sections showed a network of broad and irregularly arranged trabeculae with retained cartilage cores in the metaphyseal spongiosa.8 It contrasts with the narrow and short metaphyseal trabeculae in other lethal or severe cases of OI.5 Hypothetically, the cartilaginous trabeculae may be resistant to compression forces but susceptible to bending forces explaining the long bone distortion and absence of vertebral compression fractures in OI IIC.
The C-propeptide of type I collagen has a pivotal role in collagen assembly. Previous reports on C-propeptide mutations of COL1A1 have reported delayed trimer assembly, diminished secretion and reduced production of the total amount of procollagen.9,10,12 In contrast, it is known that the C-propeptide of type I collagen modulates TGF-beta and collagen synthesis in osteoblast cells at the early stage of differentiation.13,14 Therefore, the C-propeptide of type I collagen acts as a signaling molecule.7 This may be an explanation for the dense bone structure observed in OI IIC; however, the pathogenic mechanism remains largely unknown.
In conclusion, we report an additional case of OI IIC caused by a novel heterozygous frameshift mutation in the C-propeptide region of COL1A1. Patients with OI IIC are likely to harbor a dominant, de novo mutation of COL1A1.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at http://dx.doi.org/10.6084/m9.figshare.hgv.522.
References
References
Marini JC, Blissett AR . New genes in bone development: what's new in osteogenesis imperfecta. J Clin Endocrinol Metab 2013; 98: 3095–3103.
Sillence DO, Senn A, Danks DM . Genetic heterogeneity in osteogenesis imperfecta. J Med Genet 1979; 16: 101–116.
Spranger J . Osteogenesis imperfecta: a pasture for splitters and lumpers. (Editorial). Am J Med Genet 1984; 17: 425–428.
Thompson EM, Young ID, Hall CM, Pembrey ME . Recurrence risks and prognosis in severe sporadic osteogenesis imperfecta. J Med Genet 1987; 24: 390–405.
van der Harten HJ, Brons JT, Dijkstra PF, Meijer CJ, van Geijn HP, Arts NF et al. Perinatal lethal osteogenesis imperfecta: radiologic and pathologic evaluation of seven prenatally diagnosed cases. Pediatr Pathol 1988; 8: 233–252.
Sillence DO, Barlow KK, Garber AP, Hall JG, Rimoin DL . Osteogenesis imperfecta type II: delineation of the phenotype with reference to genetic heterogeneity. Am J Med Genet 1984; 17: 407–423.
Pace JM, Chitayat D, Atkinson M, Wilcox WR, Schwarte U, Byers PH . A single amino acid substitution (D1441Y) in the carboxyl-terminal propeptide of the proα1(I) chain of type I collagen results in a lethal variant of osteogenesis imperfecta with features of dense bone diseases. J Med Genet 2002; 39: 23–29.
Takagi M, Hori N, Chinen Y, Kurosawa K, Tanaka Y, Oku K et al. Heterozygous C-propeptide mutations in COL1A1: osteogenesis imperfecta type IIC and dense bone variant. Am J Med Genet 2011; 155: 2269–2273.
Chessler SD, Wallis GA, Byers PH . Mutations in the carboxyl-terminal propeptide of the pro alpha 1(I) chain of type I collagen result in defective chain association and produce lethal osteogenesis imperfecta. J Biol Chem 1993; 268: 18218–18225.
Lamandé SR, Chessler SD, Golub SB, Byers PH, Chan D, Cole WG et al. Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro alpha 1 (I) chain carboxyl-terminal propeptide which impair subunit assembly. J Biol Chem 1995; 270: 8642–8649.
Pace JM, Kuslich CD, Willing MC, Byers PH . Disruption of one intrachain disulphide bond in the carboxyl-terminal propeptide of the proalpha1(I) chain of type I procollagen permits slow assembly and secretion of overmodified, but stable procollagen trimers and results in mild osteogenesis imperfecta. J Med Genet 2001; 38: 443–449.
Symoens S, Hulmes DJ, Bourhis JM, Coucke PJ, Paepe AD, Malfait F . Type I . Procollagen C-propeptide defects: study of genotype-phenotype correlation and predictive role of crystal structure. Hum Mutat 2014; e-pub ahead of print 21 August 2014; 10.1002/humu.22677.
Mizuno M, Fujisawa R, Kuboki Y . Carboxyl-terminal propeptide of type I collagen (C-propeptide) modulates the action of TGF-beta on MC3T3-E1 osteoblastic cells. FEBS Lett 2000; 479: 123–126.
Mizuno M, Fujisawa R, Kuboki Y . The effect of carboxyl-terminal propeptide of type I collagen (c-propeptide) on collagen synthesis of preosteoblasts and osteoblasts. Calcif Tissue Int 2000; 67: 391–399.
Data Citations
Takagi, Masaki HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.522
Acknowledgements
We thank Prof Takao Takahashi for fruitful discussion. This study was supported by grants from the Tokyo Metropolitan Foundation (to MT).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Takagi, M., Matsushita, M., Nishimura, G. et al. Osteogenesis imperfecta IIC caused by a novel heterozygous mutation in the C-propeptide region of COL1A1. Hum Genome Var 1, 14025 (2014). https://doi.org/10.1038/hgv.2014.25
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.25
This article is cited by
-
A novel COL1A1 mutation in a family with osteogenesis imperfecta associated with phenotypic variabilities
Human Genome Variation (2017)
