
Abstract
Gorlin syndrome is an autosomal dominant disorder characterized by a wide range of developmental abnormalities and a predisposition to various tumors, and it is linked to the alteration of several causative genes, including PTCH1. We performed targeted resequencing using a next-generation sequencer to analyze genes associated with known clinical phenotypes in an 11-year-old male with sporadic jaw keratocysts. A novel duplication mutation (c.426dup) in PTCH1, resulting in a truncated protein, was identified.
Similar content being viewed by others

Basal cell nevus syndrome (BCNS; MIM #109400) or Gorlin syndrome,1 also known as nevoid basal cell carcinoma syndrome (NBCCS) or Gorlin–Goltz syndrome, is a rare autosomal dominant disorder with almost 100% penetrance and variable expressivity. Gorlin syndrome is characterized by a wide range of developmental abnormalities and a predisposition to neoplasms. Mutations in the PTCH1 gene (MIM #601309) are the main molecular defects associated with Gorlin syndrome,2,3 although mutations in other genes involved in the hedgehog pathway, such as PTCH2 (MIM #603673) and SUFU (MIM #607035),4,5 have also been reported in patients with this syndrome. Here, we report a novel PTCH1 gene duplication mutation (c.426dup) that was detected in a patient with a history of multiple keratocystic odontogenic tumors (KCOTs, Figure 1a) of the jaw, which is the most consistent and common manifestation of Gorlin syndrome.6

(a) Dental orthopantomography of a patient affected by multiple keratocystic odontogenic tumors (KCOTs). (b) Numerous tiny black pits over the right plantar area (arrows). (c) Radiograph of the skull revealing bulging of the sella turcica (arrow).
An 11-year-old Chinese male with no family history of consanguinity presented to our clinic with plantar pits (Figure 1b), mild macrocephaly, a coarse face and melanotic macules. His psychomotor development was appropriate for his age, and the results of neurological and ophthalmological examinations were normal. Imaging tests (magnetic resonance imaging and orthopantomography) revealed multiple odontogenic cysts (Figure 1a). A radiograph of the skull revealed bilamellar calcifications of the falx cerebri and bulging of the sella turcica (Figure 1c). Histopathological examination of the odontogenic cysts confirmed the diagnosis of KCOTs. Over 2 years of follow-up, new black pit lesions appeared on his right cheek. However, neither basal cell carcinoma nor bifid, fused or markedly splayed ribs were observed.
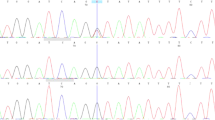
On the basis of the presence of three major criteria for clinical diagnosis,7 the patient was diagnosed with Gorlin syndrome. The molecular diagnosis was performed using genomic DNA extracted from the patient’s whole blood after obtaining informed consent. The study was approved by the ethical committees of Kobe University and The University of Tokushima. Because various alterations, including mutations and copy number alterations (CNAs)8 of three genes with numerous exons (PTCH1, PTCH2 and SUFU) can be responsible for Gorlin syndrome, we first used a Miseq bench-top sequencer (Illumina, San Diego, CA, USA) to perform next-generation sequencing with a TruSight One Sequencing Panel (Illumina) for the simultaneous targeted sequencing of the exon regions of 4,813 clinically relevant genes. The sequencing reads were aligned to the human reference genome (hg19) using Burrows–Wheeler aligner (BWA, version 0.7.8; http://bio-bwa.sourceforge.net/).9 The alignments were converted from a sequence alignment map (SAM) format to a sorted and indexed binary alignment map files (SAMtools version 0.1.19; http://samtools.sourceforge.net). Duplicate reads were removed using Picard (http://picard.sourceforge.net). Local realignment around the indels, base quality score recalibration and UnifiedGenotyper calls were performed using Genome Analysis Toolkit software (GATK, version 3.1-1; http://www.broadinstitute.org/gatk/).10 All sequence variants were annotated using Annovar.11 To identify single-nucleotide variations, we excluded sequence variants with minor allele frequency >0.05 from the 1000 Genomes Project databases (http://www.1000genomes.org/), NHLBI GO Exome Sequencing Project (ESP6500, http://evs.gs.washington.edu/EVS/) and the Human Genetic Variation Database (HGVD, http://www.genome.med.kyoto-u.ac.jp/SnpDB/). To complement the single-nucleotide variations and indel analyses, a CNA detection algorithm was applied to the alignment map files to identify large CNAs as follows: (1) binary alignment map files were converted into files covering target regions using GATK, (2) log coverage ratios and Z scores were calculated between the case and other samples and (3) regions with an abnormal copy number were detected using circular binary segmentation with DNAcopy (R/Bioconductor; http://bioconductor.org).12 These analyses identified one heterozygous frameshift mutation NM_000264.3 (PTCH1_v001):c.426dup in exon 3 of the PTCH1 gene, which caused a frameshift at codon 143, resulting in the introduction of a stop codon at position 155 (NM_000264.3 (PTCH1_i001):p.(Thr143Tyrfs*12)). This resulted in the loss of most of the coding region, which resulted in a loss of protein function; thus, this mutation is the disease-causing alteration in our patient. No other alterations, including CNAs, were observed around the three genes. The alteration was then confirmed using PCR-based direct Sanger sequencing with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) on a 3130 Genetic Analyzer (Applied Biosystems) in both directions (Figure 2, primer information is available on request). This mutation has not been previously reported in any patients with Gorlin syndrome (Human Gene Mutation Database professional 2014.2, http://www.hgmd.org/ and ClinVar, http://www.ncbi.nlm.nih.gov/clinvar/) and is not present in the 1000 Genomes, ESP6500 or HGVD databases. Because parental DNA was not available, this mutation could not be confirmed to be de novo.

Electropherogram of the PTCH1 exon 3 sequence showing the heterozygous germline duplication of a thymine (T) in the patient DNA. The DNA and corresponding amino-acid sequences of wild-type and mutant PTCH1 alleles are also shown. The affected transcript and protein were labeled NM_000264.3 (PTCH1_v001):c.426dup and NM_000264.3 (PTCH1_i001):p.(Thr143Tyrfs*12), respectively, using the Human Genome Variation Society (HGVS) nomenclature version 2.0 (Mutalyzer 2.0.beta-32, https://mutalyzer.nl/). The arrow indicates the duplication point.
The human PTCH1 gene contains 23 coding exons that span ~70 kb and encode a protein composed of 1,447 amino acids with 12 transmembrane-spanning domains and two large extracellular loops.3 PTCH1 is the ligand-binding component of the sonic hedgehog receptor complex. Aberrant activation of the sonic hedgehog signaling cascade owing to haploinsufficiency of PTCH1 is believed to cause Gorlin syndrome.13 As germline mutations of PTCH1 in patients with Gorlin syndrome were first reported by two groups in 1996,2,3 >300 different mutations covering almost all exons, with no hot spots and various CNAs, have been reported. Most of these are frameshift or nonsense mutations that lead to the synthesis of a truncated protein. In a series of 313 PTCH1 mutations reported in the Human Gene Mutation Database, most of which were detected in cases of Gorlin syndrome, 33.9% were missense or nonsense mutations, 47.6% were small insertions and/or deletions and 8.3% were gross deletions, insertions or duplications. In the current study, we reported a novel germline duplication (insertion) mutation of PTCH1, c.426dup, in a Chinese patient with sporadic Gorlin syndrome. This mutation created a premature termination codon in the mutant allele that resulted in the truncation of PTCH1. This truncation of PTCH1 is one of the shortest forms reported to date; this truncation lacks 11 of the 12 transmembrane-spanning domains. Because a premature termination codon leads to mRNA degradation via nonsense-mediated mRNA decay,14 the haploinsufficiency of PTCH1 likely had an important etiological role in this case.
A combination of clinical and molecular screening tools could be useful for the identification of Gorlin syndrome in patients who were previously diagnosed with multiple KCOTs of the jaw. These patients could be identified by retrospectively evaluating the pathological records.15 Although this case presented with several symptoms that fulfilled the criteria for the diagnosis of Gorlin syndrome, identifying the specific pathogenic mutation is useful for genetic counseling. Therefore, a proper diagnosis is essential, particularly during the early stages of life. Gorlin syndrome is primarily caused by mutations in PTCH1. However, no hot-spot mutations within the 23 coding exons have been reported in this gene; CNAs are also known to be responsible for Gorlin syndrome. Genetic alterations in PTCH2 (22 coding exons) and SUFU (12 coding exons) have also been identified, although they are rarely observed. Previous studies have revealed that there were no genotype–phenotype correlations between mutations in PTCH1 or other genes and the major clinical features of Gorlin syndrome.16,17 Target resequencing of the possible disease-causing genes and other genes associated with various known clinical phenotypes using next-generation sequencing technology enables the simultaneous evaluation of mutations and CNAs. Therefore, this could form a simple, efficient and economical genetic test for Gorlin syndrome.
References
References
Gorlin RJ, Goltz RW . Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib: a syndrome. N Engl J Med 1960; 262: 908–912.
Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85: 841–851.
Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996; 272: 1668–1671.
Fan Z, Li J, Du J, Zhang H, Shen Y, Wang CY et al. A missense mutation in PTCH2 underlies dominantly inherited NBCCS in a Chinese family. J Med Genet 2008; 45: 303–308.
Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C et al. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A 2009; 149A: 1539–1543.
Gorlin RJ . Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987; 66: 98–113.
Lo Muzio L . Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J Rare Dis 2008; 3: 32.
Fujii K, Ishikawa S, Uchikawa H, Komura D, Shapero MH, Shen F et al. High-density oligonucleotide array with sub-kilobase resolution reveals breakpoint information of submicroscopic deletions in nevoid basal cell carcinoma syndrome. Hum Genet 2007; 122: 459–466.
Li H, Durbin R . Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010; 20: 1297–1303.
Wang K, Li M, Hakonarson H . ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Olshen AB, Venkatraman ES, Lucito R, Wigler M . Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 2004; 5: 557–572.
Ingham PW, McMahon AP . Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001; 15: 3059–3087.
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE . Nonsense-mediated decay approaches the clinic. Nat Genet 2004; 36: 801–808.
Pastorino L, Pollio A, Pellacani G, Guarneri C, Ghiorzo P, Longo C et al. Novel PTCH1 mutations in patients with keratocystic odontogenic tumors screened for nevoid basal cell carcinoma (NBCC) syndrome. PLoS ONE 2012; 7: e43827.
Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S et al. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype–phenotype correlations are evident. Am J Hum Genet 1997; 60: 21–26.
Bale AE, Gailani MR, Leffell DJ . The Gorlin syndrome gene: a tumor suppressor active in basal cell carcinogenesis and embryonic development. Proc Assoc Am Phys 1995; 107: 253–257.
Data Citations
Imoto, Issei HGV Database (2014) http://dx.doi.org/10.6084/m9.figshare.hgv.513
Acknowledgements
We thank the patient and his family for their participation in this study. We also thank Dr Hiroshi Nagai for his technical assistance. This work was supported by JSPS KAKENHI Grant Numbers 60645216 (NO) and 26293304 (II) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Okamoto, N., Naruto, T., Kohmoto, T. et al. A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Var 1, 14022 (2014). https://doi.org/10.1038/hgv.2014.22
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.22
This article is cited by
-
Functionally confirmed compound heterozygous ADAM17 missense loss-of-function variants cause neonatal inflammatory skin and bowel disease 1
Scientific Reports (2021)
-
Molecular diagnosis of an infant with TSC2/PKD1 contiguous gene syndrome
Human Genome Variation (2020)
-
Novel compound heterozygous CDH23 variants in a patient with Usher syndrome type I
Human Genome Variation (2019)
-
Primary microcephaly caused by novel compound heterozygous mutations in ASPM
Human Genome Variation (2018)
-
The first Japanese patient with mandibular hypoplasia, deafness, progeroid features and lipodystrophy diagnosed via POLD1 mutation detection
Human Genome Variation (2017)
