
Abstract
Familial hypercholesterolemia (FH) is an autosomal dominant disease predominantly caused by a mutation in the low-density lipoprotein receptor (LDLR) gene. Here, we describe two severely affected FH patients who were resistant to statin therapy and were managed on an apheresis program. We identified a novel duplication variant c.1332dup, p.(D445*) at exon 9 and a known silent variant c.1413A>G, p.(=), rs5930, NM_001195798.1 at exon 10 of the LDLR gene in both patients.
Similar content being viewed by others


Introduction
Familial hypercholesterolemia (FH) is an autosomal dominant disorder that predisposes the patient to the development of coronary artery disease and sudden cardiac death. FH is generally caused by mutations in the low-density lipoprotein receptor (LDLR) gene, leading to reduced hepatic clearance of LDL from the blood. FH can also be caused by mutations in the apolipoprotein B (APOB) gene, which encodes the LDLR ligand. In addition, a pathogenic mutation in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene has been proposed to cause FH, indicating that the disease is genetically heterogeneous.1–3 Several studies have demonstrated that mutations in the LDLR, APOB or PCSK9 genes can result in hypercholesterolemia.
The LDLR gene consists of 18 exons spanning 45 kb and encodes an 860-amino-acid precursor protein.4 In the majority of populations, the frequency of heterozygosity is less than 1:500, whereas the homozygous form is rare, occurring at a frequency of 1:1,000,000.5 Worldwide, there are more than 1,288 reported genetic variants in LDLR.6–8 The highest frequency of heterozygosity is observed in the South African population, with an incidence of less than 1:80.9 Other studies in the French-Canadian population found five common variants with a frequency of 1:270.4,10 This high frequency is attributed to the founder effect and consanguineous marriages.
Before this research, a genetic epidemiological study of the frequency of FH in the Saudi population had not been conducted. We expect the incidence of FH, secondary to the homozygous genotype in Saudi Arabia, to be relatively high, because of the increased rate of consanguineous marriages (over 54%). Consequently, many cardiovascular centers have been established across the country to provide adequate care and treatment for patients with heart diseases.11 The estimated number of affected heterozygote Saudi individuals ranges from 46,000 to 230,000 individuals. The majority of affected individuals may not be aware of the disease, as they remain asymptomatic until a severe myocardial infarction occurs, usually after the age of 40 years. The myocardial infarctions can be severe enough to cause sudden cardiac death or disabling cardiovascular morbidities. For the clinical diagnosis of FH, there are three sets of criteria used: the Simon Broome Register (UK), the Dutch Lipid Clinic Network (the Netherlands) and the MEDPED Program (USA). These criteria have been proven to identify FH in patients. The criteria are primarily based on age, blood cholesterol levels and evidence of clinical signs related to FH (xanthelasma, tendinous xanthomata and corneal arcus) and a family history of coronary heart disease (CHD). However, elevated blood cholesterol levels are commonly observed in individuals with nongenetic multifactorial hypercholesterolemia, which may lead to a misdiagnosis of FH in those individuals. Therefore, the use of molecular methods to characterize gene defects is necessary for an unequivocal FH diagnosis. Furthermore, when novel variants are identified, distinguishing between pathogenic variants and polymorphs is crucial for molecular confirmation. Further investigation into the pathogenicity of novel variants can be performed by standard functional analyses or by using new alternative bioinformatics methods to predict the putative effects of variants on protein function and stability.12
Materials and methods
Subjects
The analysis was performed in two patients diagnosed with homozygous FH. The patients are siblings whose family originates from a tribe that lives in the northern region of Saudi Arabia. Both patients are resistant to statin therapy and were on an apheresis program. Sample collection and studies were performed in accordance with the Research Ethics Committee’s regulation after the subjects were provided with informed consent. The enrollment criteria for the genetic screening of the patients were based on the Simon Broome register.13
DNA analysis
Genomic DNA was isolated from EDTA-treated whole blood using the MagNA Pure Compact Nucleic Acid Isolation Kit I (Roche, Basel, Switzerland) according to the manufacturer’s instructions. Polymerase chain reaction (PCR) amplification of the LDLR gene (including the 18 coding exons and flanking intron regions), APOB gene (exon 26 of the APOB gene containing codons 3,475–3,592, which harbors three known pathogenic variant sites, R3500Q, R3500W and R3527Q) and the PCSK9 gene (the 12 exons and flanking intron regions) were performed. Descriptions of the primers used for amplifying and sequencing the fragments are provided in Supplementary Table 1. PCR was performed with 100 ng genomic DNA using the HotStarTaq Plus DNA Polymerase Kit (Qiagen, Hilden, Germany) as follows: Taq polymerase was activated at 94 °C for 5 min, followed by 35 cycles of denaturing at 94 °C for 30 s, annealing at 61–64 °C (Supplementary Table 1) for 30 s, extension at 72 °C for 45 s and final extension at 72 °C for 5 min. The amplified products were separated on an agarose gel to ensure the size and quality of the band. The PCR products were purified using magnetic beads with the Agencourt AMPure XP kit (Beckman Coulter, Brea, CA, USA). The purified products were used as templates for direct sequencing with a BigDye Terminator v3.1 cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA). The sequencing reaction products were purified using the BigDye X-terminator purification kit (Applied Biosystems) followed by capillary electrophoresis in an ABI 3500 Genetic analyzer (Applied Biosystems). The final analysis was performed using the Sequence Analysis Software v5.4 (Applied Biosystems).
Results
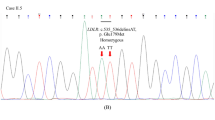
The genomic analysis revealed a novel duplication variant c.1332dup, p.(D445*) at exon 9 of the LDLR gene in the two patients (Figure 1). Both patients were determined to be homozygous for this variant, which expresses a premature stop codon at position 445 in exon 9 of the LDLR gene. Consequently, a truncated protein is produced, which is a defective LDL receptor. In addition to this novel nonsense variant, we also identified a known silent variant c.1413A>G, p.(=), rs5930, NM_001195798.1 in both patients at exon 10 of the LDLR gene (Table 1). Figure 1a shows the pedigree of the family (the parents and the first-degree relatives were not available for DNA analysis at the time of the study). Table 1 also shows the total and LDL cholesterol levels with values reaching 18.11 and 15.05 mmol/l in patient one and 15.13 and 12.98 mmol/l in patient two. These values are considered very high compared with the optimal level, which is <2.59 mmol/l (LDL cholesterol). This confirms the loss of LDLR function because of the nonsense variant present in the DNA sequence.

Novel nonsense variant p.(D445*) identified at exon 9 of the LDLR gene. (a) Pedigree of the patient family. (b) Representative DNA sequence from the control individuals (wild type; WT) and patients. ?=NOT available for DNA analysis. *=Index case.
Discussion
To date, there are more than 1,288 LDLR variants reported in FH patients worldwide that are regarded as pathogenic variants.8,14–16 We identified a novel duplication variant (c.1332dup) at exon 9 of the LDLR gene. This duplication generates a defective LDL receptor because of the premature stop codon at position 445 in exon 9 of the human LDLR gene. No other variants were observed in the LDLR, APOB and PCSK9 genes.
This novel nonsense variant is localized in the EGF precursor homology domain. Theoretically, the membrane-spanning domain and the remainder of the LDLR protein cannot be synthesized; therefore, the truncated protein may be degraded by the proteasome machinery. This hypothesis agrees with the patient phenotypes, that is, both patients were resistant to statin therapy and were on an apheresis program. Furthermore, an earlier study determined that up to 54% of LDLR variants that resulted in FH were localized in the epidermal growth factor precursor (EGFP) homology domain.16 Marduel et al.7 reported nine nonsense variants that were classified as FH-causing variants because of the synthesis of a truncated protein. In addition, the lipid profile of our patients supports the loss of LDLR function, as the analysis of the total and LDL cholesterol levels revealed an extremely high concentration of more than 12 mmol/l. Interestingly, the levels of LDL and total cholesterol in both our patients are higher compared with the maximal level of the FH group that was presented in Marduel et al.7 This suggests that it is necessary to establish a validated LDL concentration cut-off for the clinical diagnosis of FH in Saudi Arabia. In conclusion, the segregation pattern of the variant is consistent with the lipid profile (Figure 1 and Table 1), suggesting a more severe FH phenotype when the variant is in the homozygous state.
References
Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34: 154–156.
Timms KM, Wagner S, Samuels ME, Forbey K, Goldfine H, Jammulapati S et al. A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet 2004; 114: 349–353.
Al-Allaf FA, Coutelle C, Waddington SN, David AL, Harbottle R, Themis M . LDLR-Gene therapy for familial hypercholesterolaemia: problems, progress, and perspectives. Int Arch Med 2010; 3: 36.
Hobbs HH, Brown MS, Goldstein JL . Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992; 1: 445–466.
Heath KE, Humphries SE, Middleton-Price H, Boxer M . A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur J Hum Genet 2001; 9: 244–252.
Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE . Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet 2008; 72: 485–498.
Marduel M, Carrie A, Sassolas A, Devillers M, Carreau V, Di Filippo M et al. Molecular spectrum of autosomal dominant hypercholesterolemia in France. Hum Mutat 2010; 31: E1811–E1824.
Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C et al. Low-density lipoprotein receptor gene familial hyperc- holesterolemia variant database: update and pathological assess- ment. Ann Hum Genet 2012; 76: 387–401.
Steyn K, Goldberg YP, Kotze MJ, Steyn M, Swanepoel AS, Fourie JM et al. Estimation of the prevalence of familial hypercholesterolaemia in a rural Afrikaner community by direct screening for three Afrikaner founder low density lipoprotein receptor gene mutations. Hum Genet 1996; 98: 479–484.
Leitersdorf E, Tobin EJ, Davignon J, Hobbs HH . Common low-density lipoprotein receptor mutations in the French Canadian population. J Clin Invest 1990; 85: 1014–1023.
Jaber L, Halpern GJ, Shohat M . The impact of consanguinity worldwide. Community Genet 1998; 1: 12–17.
Thusberg J, Vihinen M . Pathogenic or not? And if so, then how? Studying the effects of missense mutations using bioinformatics methods. Hum Mutat 2009; 30: 703–714.
Scientific Steering Committee on behalf of the Simon Broome Register Group. Risk of fatal coronary heart disease in familial hypercholesterolaemia. Br Med J 1991; 303: 893–896.
Mollaki V, Progias P, Drogari E . Novel LDLR variants in patients with familial hypercholesterolemia: in silico analysis as a tool to predict pathogenic variants in children and their families. Ann Hum Genet 2013; 77: 426–434.
Ahmed W, Whittall R, Riaz M, Ajmal M, Sadeque A, Ayub H et al. The genetic spectrum of familial hypercholesterolemia in Pakistan. Clin Chim Acta 2013; 421: 219–225.
Wang H, Xu S, Sun L, Pan X, Yang S, Wang L . Functional characterization of two low-density lipoprotein receptor gene mutations in two chinese patients with familial hypercholesterolemia. PLoS ONE 2014; 9: e92703.
Acknowledgements
We acknowledge the patients and their family members for their cooperation. We thank the members of the Science and Technology Unit at Umm Al-Qura University for their support. We also thank Dr Lisandra De Castro Brás, University of Mississippi Medical Center, for proofreading the manuscript. This work was supported by the National Science, Technology and Innovation Plan (grant 08-BIO34-10), Kingdom of Saudi Arabia.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information for this article can be found on the Human Genome Variation website (http://www.nature.com/hgv)
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article

Cite this article
Al-Allaf, F., Athar, M., Abduljaleel, Z. et al. Identification of a novel nonsense variant c.1332dup, p.(D445*) in the LDLR gene that causes familial hypercholesterolemia. Hum Genome Var 1, 14021 (2014). https://doi.org/10.1038/hgv.2014.21
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.21
