
Abstract
Using a next-generation sequencing strategy, we identified a novel KAL1 missense mutation (p.His568Gln) in a patient with combined pituitary hormone deficiency, right microphthalmia, right renal aplasia and severe developmental delay. Our findings will provide additional evidence that KAL1 mutations are associated with hypopituitarism, in addition to luteinizing hormone, and follicle-stimulating hormone deficiencies, and improve our understanding of the phenotypic features and developmental course associated with KAL1 mutations.
Similar content being viewed by others


Kallmann syndrome (KS) is a genetically heterogeneous condition, defined by hypogonadotropic hypogonadism (HH) and anosmia/hyposmia. Several genes have been linked to KS pathogenesis, including FGFR1, FGF8, PROK2, PROKR2, CHD7 and KAL1.1–6 Increasing evidence shows that overlapping genotypes/phenotypes exist between KS, combined pituitary hormone deficiency (CPHD) and septo-optic dysplasia (SOD), a condition characterized by pituitary hormone deficiencies, optic nerve hypoplasia and midline defects, including agenesis of the septum pellucidum and/or corpus callosum. Mutations in FGFR1, FGF8 and PROKR2, the genes responsible KS, have been identified in a small number of CPHD/SOD, suggesting that the genetic overlap between KS, CPHD and SOD is significant.7–10 Unlike FGFR1, FGF8 and PROKR2, the contribution of KAL1, which is mutated in 5% of KS cases,11 to CPHD/SOD development has not been clearly established.
Here, we report a case of CPHD patient with extra-pituitary phenotypes, including right microphthalmia, right renal aplasia, mild hearing impairment in both ears and severe developmental delay. Using a next-generation sequencing strategy, we identified a novel missense mutation in KAL1 (p.His568Gln). Our findings provide additional evidence that KAL1 mutations are associated with hypopituitarism, in addition to luteinizing hormone (LH), and follicle-stimulating hormone (FSH) deficiencies, and will further our understanding of the phenotypic features, and developmental course associated with KAL1 mutations.
The propositus was a 13-year-old Japanese boy born at 40 weeks of gestation after an uncomplicated pregnancy and delivery. He had no family history of pituitary dysfunction. His parents were nonconsanguineous and phenotypically normal. The patient’s birth weight, length and head circumference were 2638 g (below the 3rd percentile), 46.0 cm (3rd–10th percentile) and 31.8 cm (3rd–10th percentile), respectively. A constellation of malformations was noticed, including right microphthalmia and small and uplifted earlobes with very-small external auditory canals. The testes were undescended, the scrotum small and the foreskin hypoplastic. Echography revealed right renal aplasia. An auditory brainstem response examination revealed mild hearing impairment in both ears. Owing to severe psychomotor retardation, he remains wheelchair-bound and nonverbal at 13 years of age.
Frequent episodes of hypoglycemia were noted at the age of 3 months. He was diagnosed with central adrenal insufficiency based on low cortisol (2.2 μg/dl) and adrenocorticotropin (ACTH) (32 pg/ml) at a time of severe hypoglycemia (glucose 1.3 mmol/l). Further endocrine studies indicated that the patient also had central hypothyroidism on the basis of a low free T4 (0.50 ng/dl: Ref. 0.99–1.91) with an inadequately increased thyroid-stimulating hormone (TSH) level of 3.98 mU/l (Ref. 0.77–7.3), and growth hormone (GH) deficiency (Supplementary Table 1). The brain MRI exhibited anterior pituitary hypoplasia, visible but thin stalk, cerebellar hypoplasia and eutopic posterior pituitary. The olfactory bulb was difficult to identify. Replacement therapy with L-thyroxine, hydrocortisone and recombinant human GH was started at 3 months of age. At 13 years of age, he showed typical signs of hypogonadism, with small intrascrotal testes (1 ml), no pubic hair (Tanner stage 1) and a micropenis (stretched penile length 2.0 cm). Hormone assays revealed very-low plasma testosterone levels. The HH diagnosis was confirmed by LH-releasing hormone stimulating test (Supplementary Table 1).
After obtaining informed consent, and with the approval of the Institutional Review Board of Keio University School of Medicine, and the Institutional Review Board of Tokyo Metropolitan Children’s Medical Center, genomic DNA was extracted from peripheral blood leucocytes of the propositus. We sequenced 9 genes implicated in CPHD, including POU1F1, PROP1, LHX3, LHX4, HESX1, OTX2, SOX3, SOX2, GLI2, and 12 genes implicated in KS/HH, including CHD7, FGFR1, FGF8, GNRH1, GNRHR, KISS1, KISS1R, PROK2, PROKR2, TAC3, TACR3 and KAL1 using the MiSeq instrument (Illumina, San Diego, CA, USA) according to the SureSelect protocol (Agilent Technologies, Santa Clara, CA, USA). In brief, 3 μg of genomic DNA was used for the SureSelect capture methods. Exons of 122 genes known to be associated with congenital endocrine disorders (including 9 CPHD and 12 KS/HH-related genes) were identified in the University of California Santa Cruz table browser (http://genome.ucsc.edu/). In total, we targeted 1321 regions comprising 2 46 158 bp using SureSelect. DNA obtained from the SureSelect solution-based sequence capture was subjected to MiSeq sequencing. Base calling, read filtering and demultiplexing were performed with the standard Illumina processing pipeline. We used BWA 0.6.1 and SAMtools 0.1.18 for alignment and variant detection against the human reference genome (NCBI build 37; hg19) with the default settings. Local realignment, quality score recalibration and variant calling were performed by GATK 2.3-9 with the default settings. We used ANNOVAR for annotation of called variants. As for LHX3 and SOX3, we screened by PCR and direct sequencing.
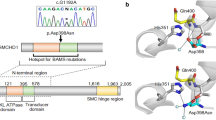
We identified a novel hemizygous KAL1 mutation, c.1704C>A (p.His568Gln), the only gene among 9 CPHD and 12 KS/HH-related genes for which unknown variants were identified. We performed Sanger sequencing on PCR products from genomic DNA to confirm the KAL1 variant (Figure 1a). The p.His568Gln was not detected in 150 healthy Japanese controls and was absent from database, including dbSNP, the 1000 Genomes Project, Exome Variant Server, NHLBI Exome Sequencing Project and the Human Genetic Variation Database in Japanese. The p.His568Gln mutation in KAL1 was submitted to in silico analysis. The results exhibited that this mutation was predicted to cause functional damage by PolyPhen-2 http://genetics.bwh.harvard.edu/pph2/ (damage score 0.928, sensitivity 0.81 and specificity 0.94). Parental analysis was refused. KAL1 encodes anosmin-1 (680 amino acids), a protein that consists of a whey acidic protein-like domain followed by four fibronectin type III (FnIII) domains. His568 is a highly evolutionarily conserved amino acid across mammals, located in the fourth FnIII domain (Figures 1a and b).

Identification of sequence variation of KAL1. (a) Partial sequence of PCR product and schematic diagrams of the anosmin-1 (coded by KAL1 gene) protein. The chromatogram represents a hemizygous substitution of glutamine (CAA) in place of histidine (CAC) at codon 568, located in the fourth FnIII domain. The arrow indicates the mutated nucleotide. The reported 3 missense mutations identified in CPHD/SOD patients are summarized. (b) His568 is a highly evolutionarily conserved amino acid across mammals.
Genomic DNAs were also subjected to array comparative genomic hybridization with the Agilent 4×180 K SurePrint G3 Human CGH Microarray (catalog no. G4449A; Agilent Technologies). No significant copy-number changes were identified.
Anosmin-1 has a key role in the migration of gonadotropin-releasing hormone neurons and olfactory nerves to the hypothalamus and olfactory bulbs, and is involved in FGF signaling, which has a positive role in pituitary cell proliferation.12,13 To date, only four CPHD/SOD patients harboring KAL1 mutations have been reported.7,14 Raivio et al.7 reported one male CPHD patient (GH, TSH, ACTH and LH/FSH deficiencies) carrying a KAL1 mutation (p.His459Tyr). However, this patient also carried a heterozygous p.Arg85His mutation in PROKR2, which had been reported to be causative for KS. Therefore, the pure contribution of the KAL1 mutation to this CPHD phenotype is not clear. McCabe et al.14 reported three female patients with SOD harboring heterozygous KAL1 mutations among 421 CPHD/SOD patients examined. Although these 421 patients were only screened for KAL1, this result implies that KAL1 mutations only contribute to CPHD/SOD etiology to a minor extent, if any. In Table 1, we summarize the clinical phenotypes and MRI findings of KAL1 mutations within CPHD/SOD patients reported to date. We believe that our findings contribute additional evidence that KAL1 mutations are associated with hypopituitarism, and provide clinical information to expand the phenotypic spectrum of patients harboring KAL1 mutations.
We used a next-generation sequencing strategy to analyze 122 genes associated with congenital endocrine disorders. The genetic etiologies of CPHD and KS/HH are quite heterogeneous, and recent studies have showed that KS/HH is not strictly a monogenic Mendelian disease as previously thought; instead, it is emerging as a digenic or potentially oligogenic disease.15,16 When multiple genes need to be analyzed simultaneously for mutations, targeted sequence analysis of interesting genomic regions is an attractive approach.
Relative to the four previously described CPHD/SOD patients carrying KAL1 mutations, our patient had severe pituitary (GH, TSH and ACTH deficiencies in addition to LH/FSH deficiencies) and extra-pituitary phenotypes, including ocular malformation as well as severe growth and psychomotor retardation. Severe HH and predominantly right-sided renal aplasia are consistent with a previously reported phenotype of a male patient harboring KAL1 mutations;17 however, the phenotypical variation could be partly due to the impact of other genes that are important, but have not yet been recognized as genes involved in pituitary development. Therefore, further studies are necessary to clarify the independent contribution of KAL1 mutations to the development of CPHD.
In conclusion, we identified a novel KAL1 mutation in a CPHD patient with extra-pituitary phenotypes, including a right microphthalmia, right renal aplasia, mild hearing impairment in both ears and severe developmental delay. This study expands our understanding of the phenotypic features and developmental course associated with KAL1 mutations.
References
References
Legouis R, Hardelin JP, Levilliers J, Claverie JM, Compain S, Wunderle V et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 1991; 67: 423–435.
Dodé C, Levilliers J, Dupont JM, De Paepe A, Le Dû N, Soussi-Yanicostas N et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet 2003; 33: 463–465.
Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2006; 2: 1648–1652.
Ogata T, Fujiwara I, Ogawa E, Sato N, Udaka T, Kosaki K . Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr J 2006; 53: 741–743.
Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 2008; 118: 2822–2831.
Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome – the clinical overlap with CHARGE syndrome. Clin Genet 2009; 75: 65–71.
Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab 2012; 97: E694–E699.
Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM et al. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab 2012; 97: E1068–E1073.
McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O et al. Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo-optic dysplasia. J Clin Endocrinol Metab 2013; 98: E547–E557.
Fukami M, Iso M, Sato N, Igarashi M, Seo M, Kazukawa I et al. Submicroscopic deletion involving the fibroblast growth factor receptor 1 gene in a patient with combined pituitary hormone deficiency. Endocr J 2013; 60: 1013–1020.
Georgopoulos NA, Pralong FP, Seidman CE, Seidman JG, Crowley Jr WF, Vallejo M . Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab 1997; 82: 213–217.
Dodé C, Hardelin JP. Kallmann syndrome: fibroblast growth factor signaling insufficiency? J Mol Med 2004; 82: 725–734.
Norlin S, Nordström U, Edlund T . Fibroblast growth factor signaling is required for the proliferation and patterning of progenitor cells in the developing anterior pituitary. Mech Dev 2000; 96: 175–182.
McCabe MJ, Hu Y, Gregory LC, Thankamony A, Hughes L, Townshend S et al. Novel KAL1 sequence variants associated with septo-optic dysplasia (SOD) in three female patients. Horm Res 2012; 78S1: 111.
Pitteloud N, Durrani S, Raivio T, Sykiotis GP . Complex genetics in idiopathic hypogonadotropic hypogonadism. Front Horm Res 2010; 39: 142–153.
Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA 2010; 34: 15140–15144.
Sato N, Katsumata N, Kagami M, Hasegawa T, Hori N, Kawakita S et al. Clinical assessment and mutation analysis of Kallmann syndrome 1 (KAL1) and fibroblast growth factor receptor 1 (FGFR1, or KAL2) in five families and 18 sporadic patients. J Clin Endocrinol Metab 2004; 89: 1079–1088.
Data Citations
Hasegawa, Tomonobu HGV Database http://dx.doi.org/10.6084/m9.figshare.hgv.505 (2014)
Acknowledgements
This work was supported by a Grant-in-Aid for the Health Science Research Grant for Research on Applying Health Technology (Jitsuyoka (Nanbyo)-Ippan-014(23300102)) from the Ministry of Health, Labour and Welfare of Japan, and grants from Tokyo Metropolitan Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Takagi, M., Narumi, S., Hamada, R. et al. A novel KAL1 mutation is associated with combined pituitary hormone deficiency. Hum Genome Var 1, 14011 (2014). https://doi.org/10.1038/hgv.2014.11
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2014.11
